Disrupting the Btk Pathway Suppresses COPD-Like Lung Alterations in Atherosclerosis Prone ApoE−/− Mice Following Regular Exposure to Cigarette Smoke

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Results
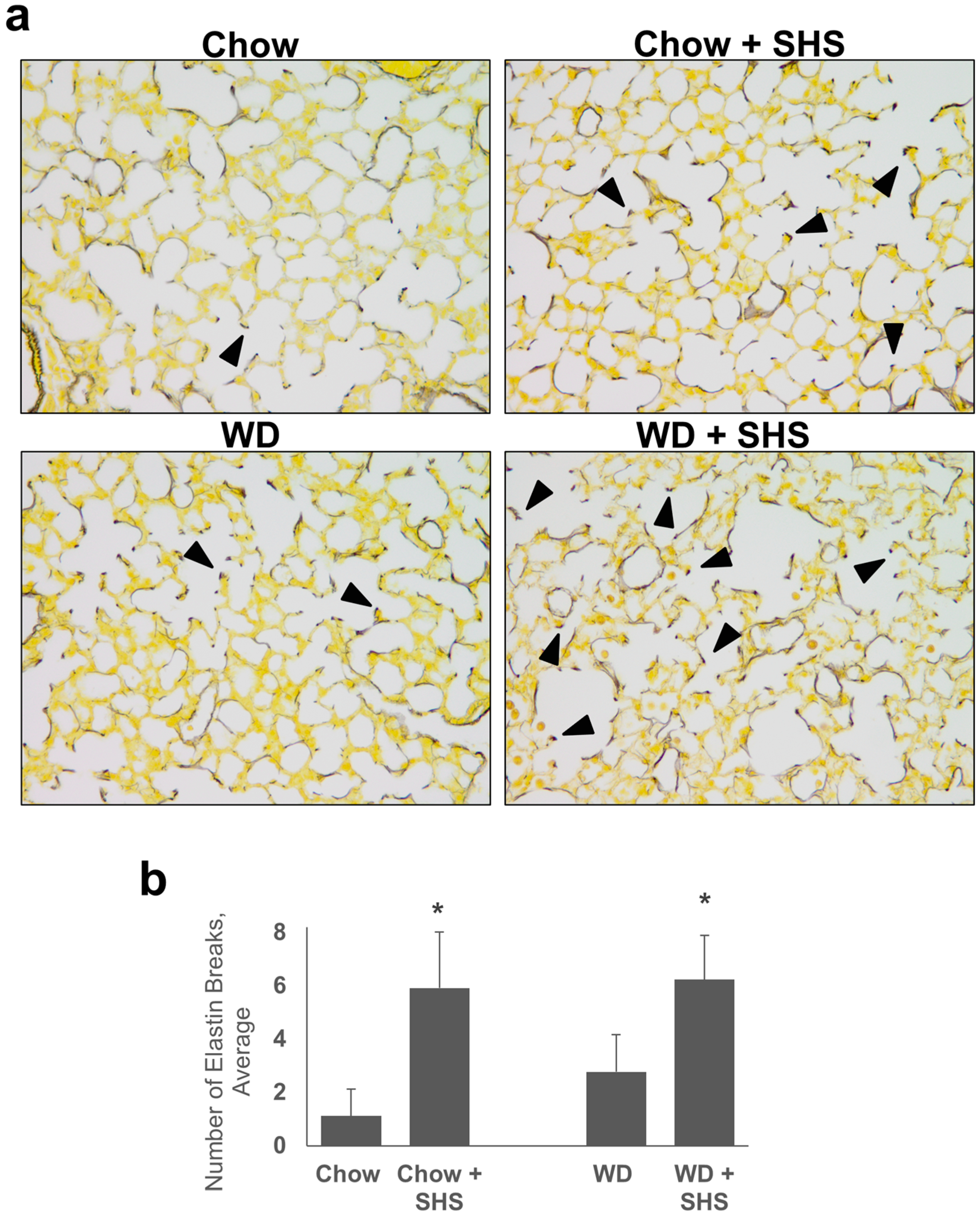
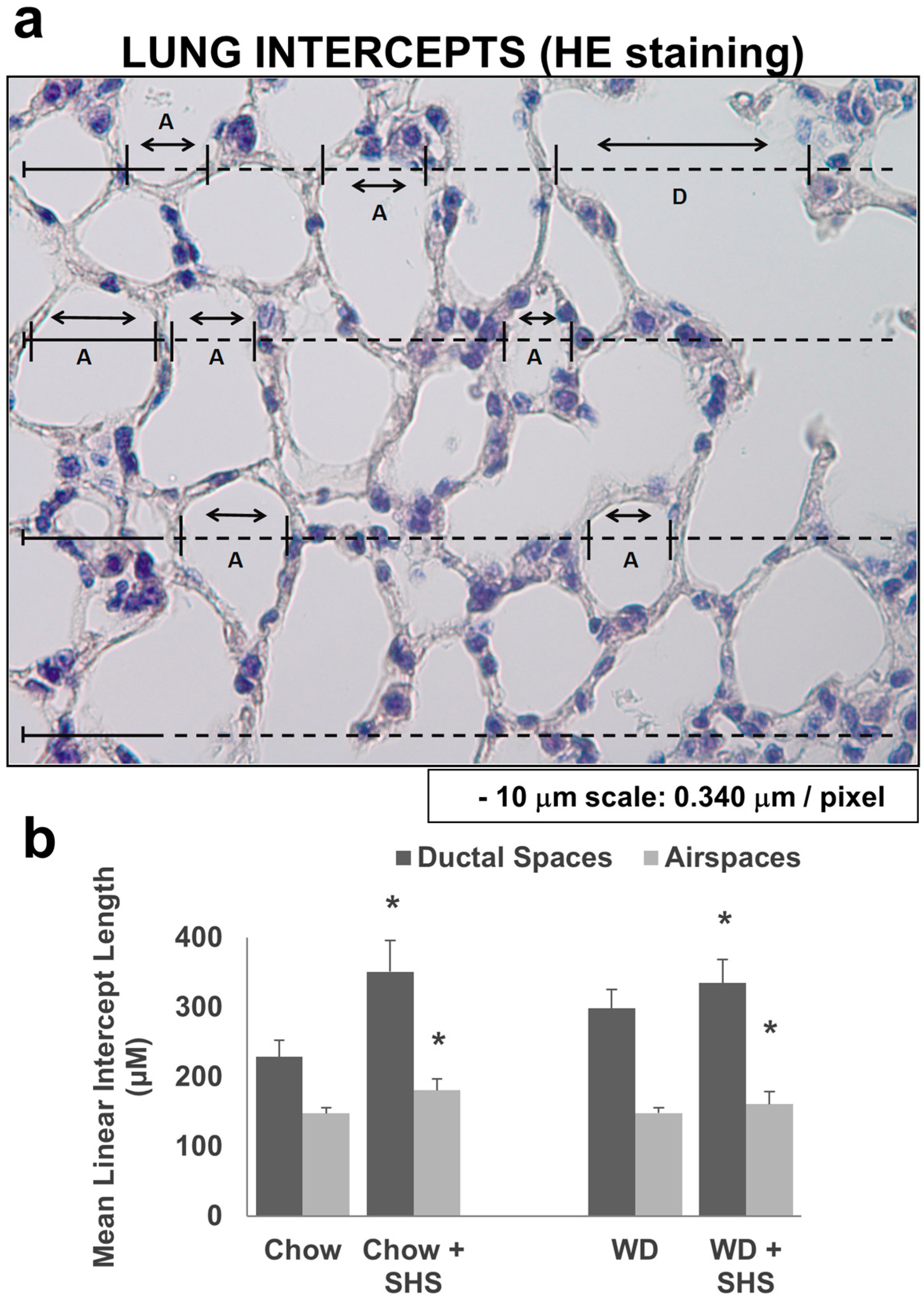
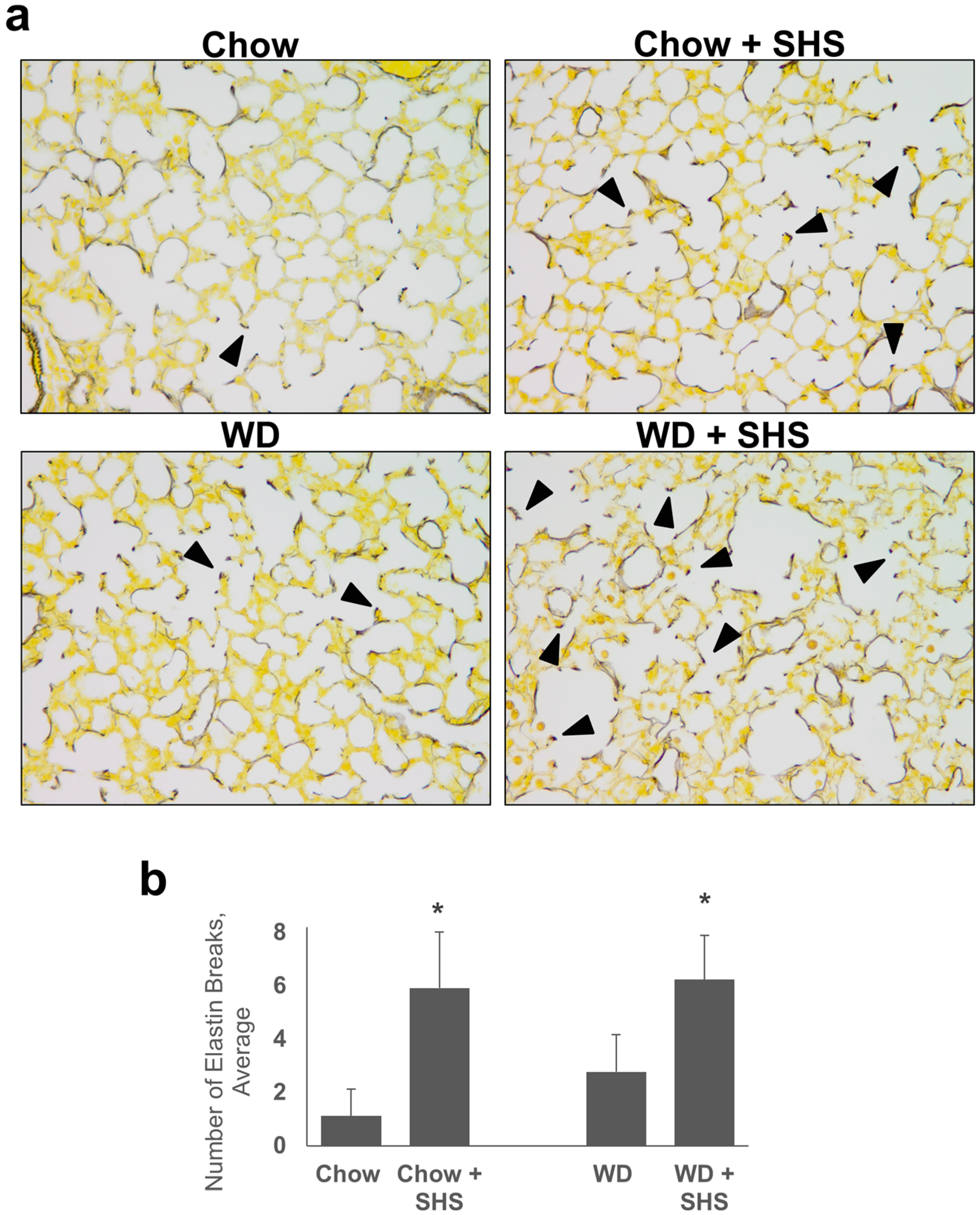
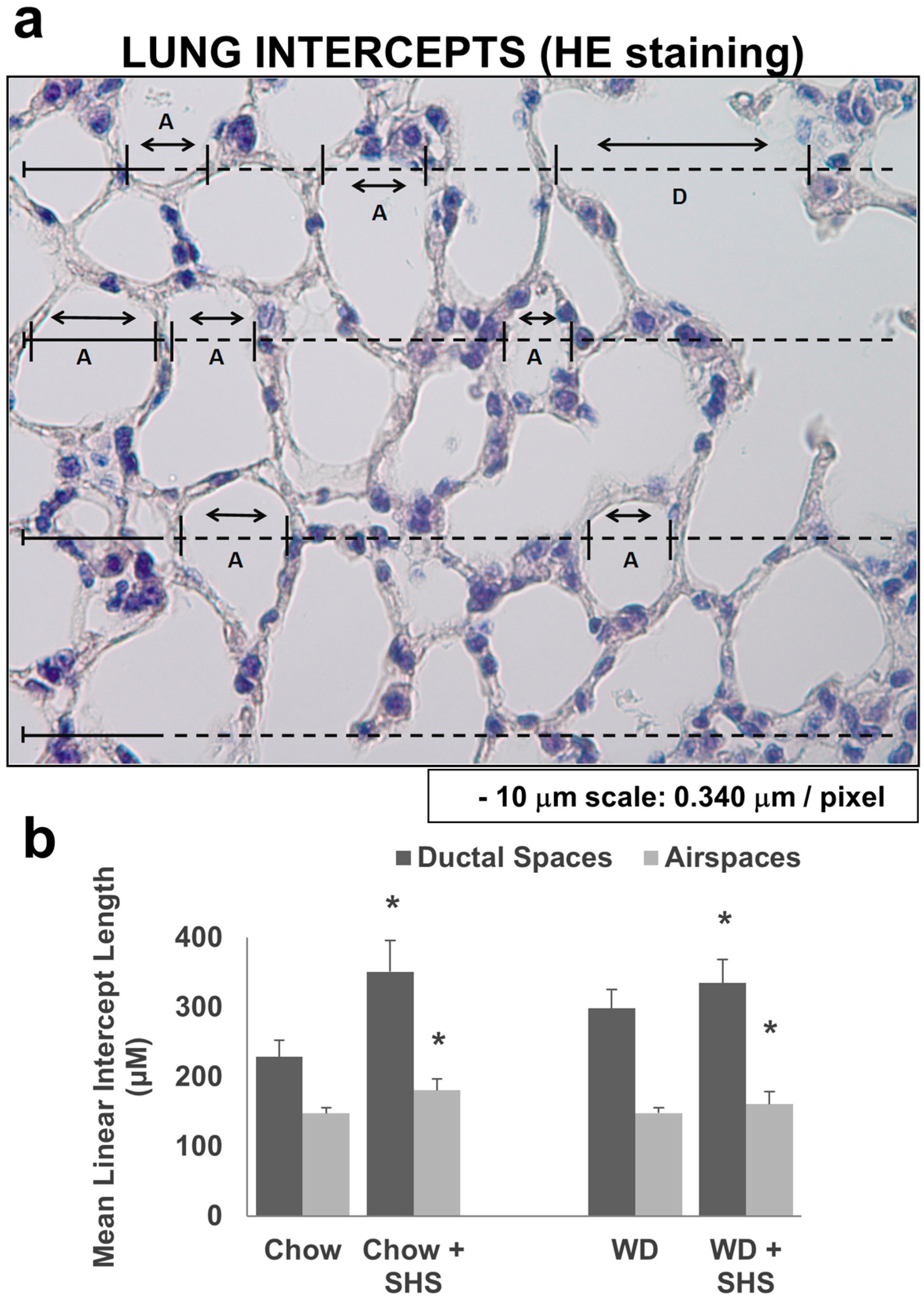
2.1. Exposure to SHS Induces Alveolar Destruction and Airspace Enlargement in Lungs of ApoE−/− Mice
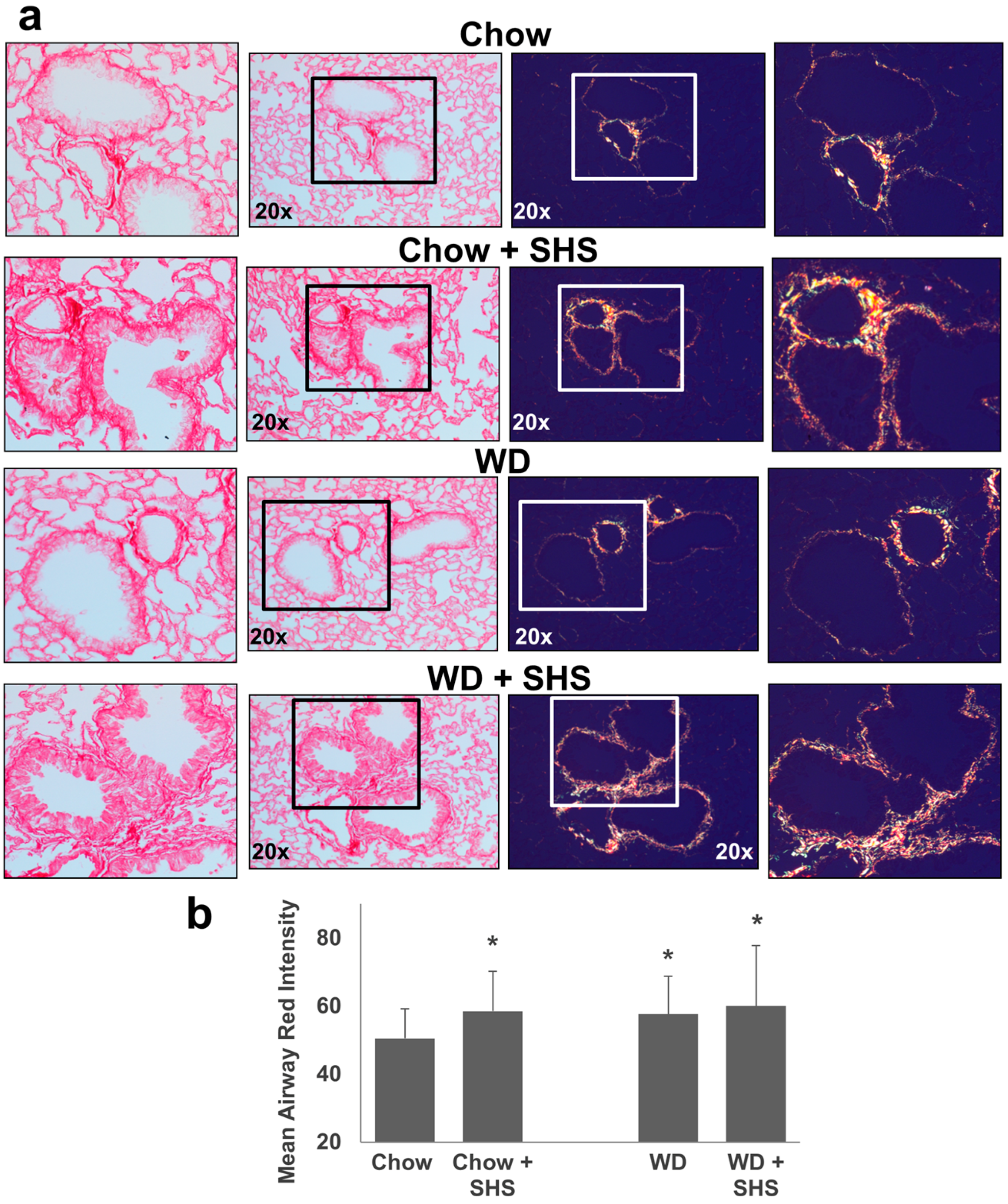
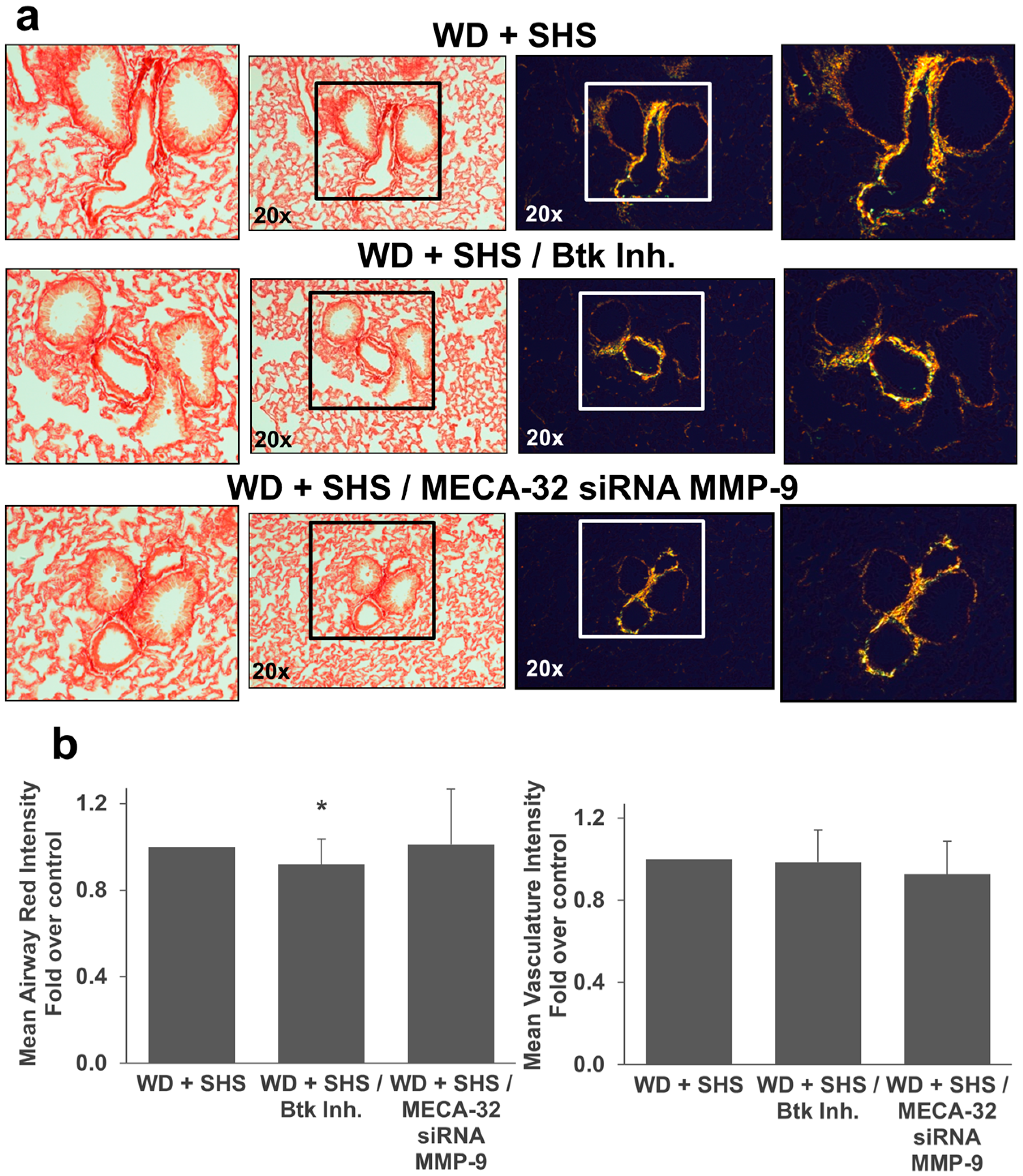
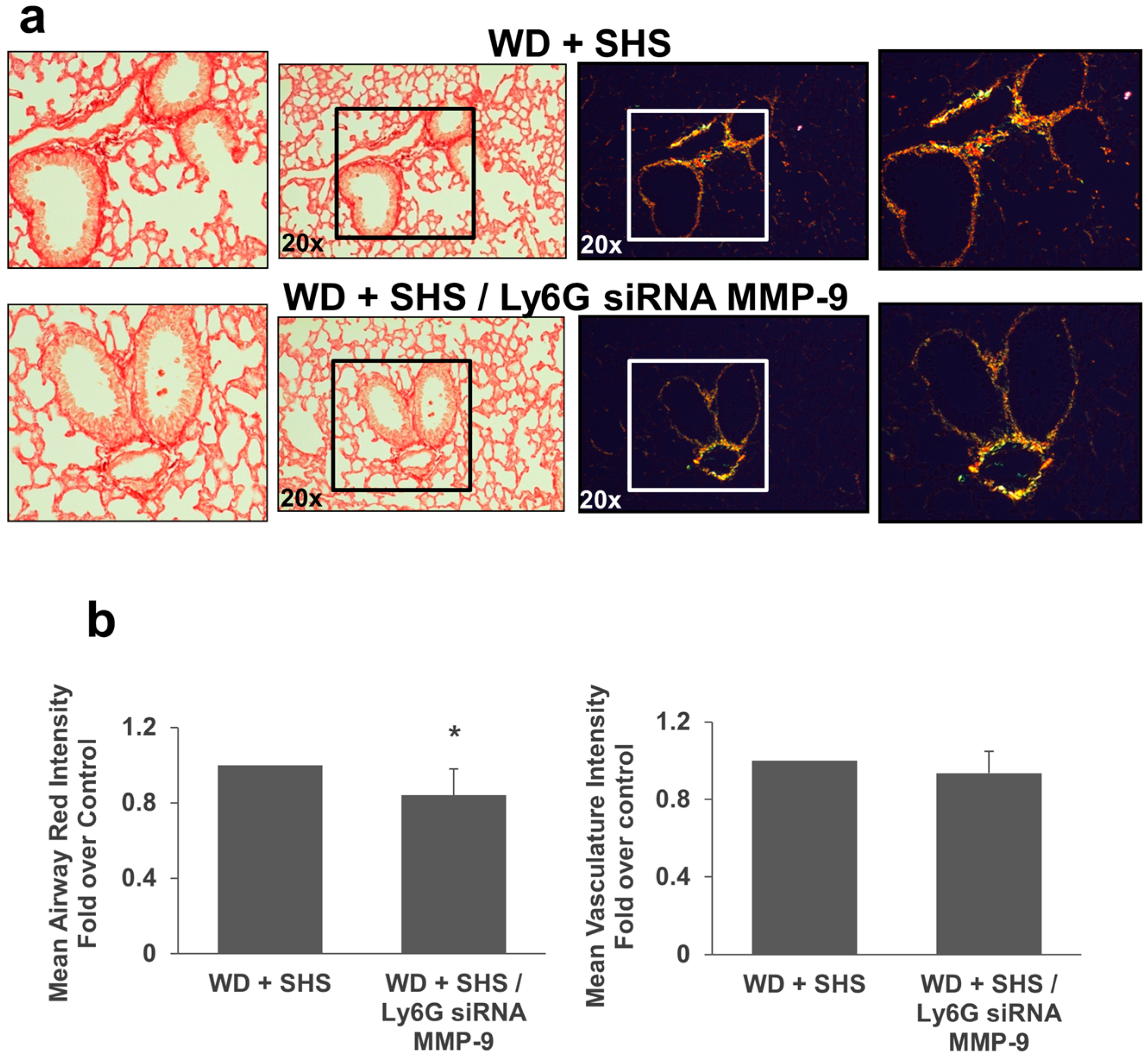
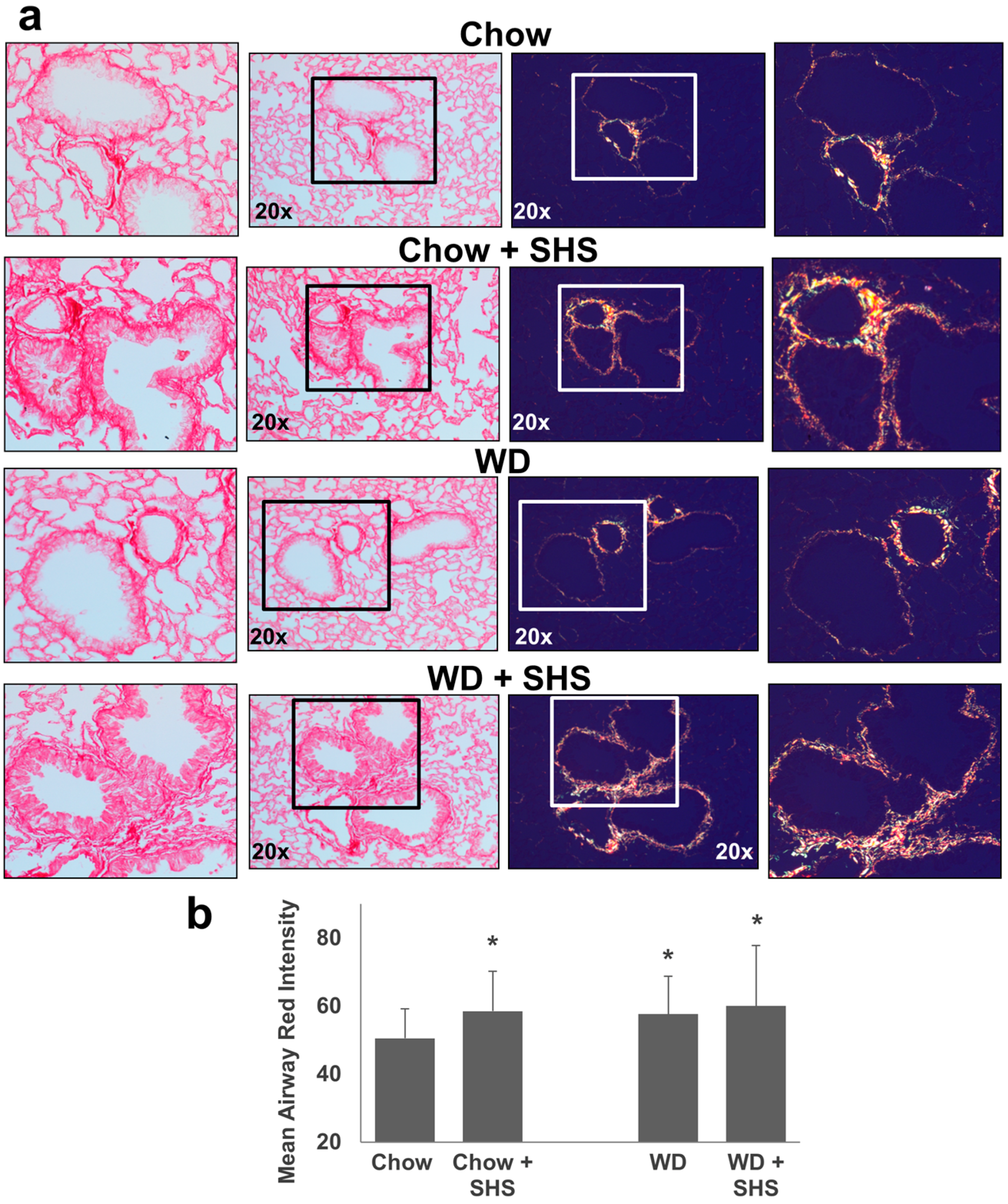
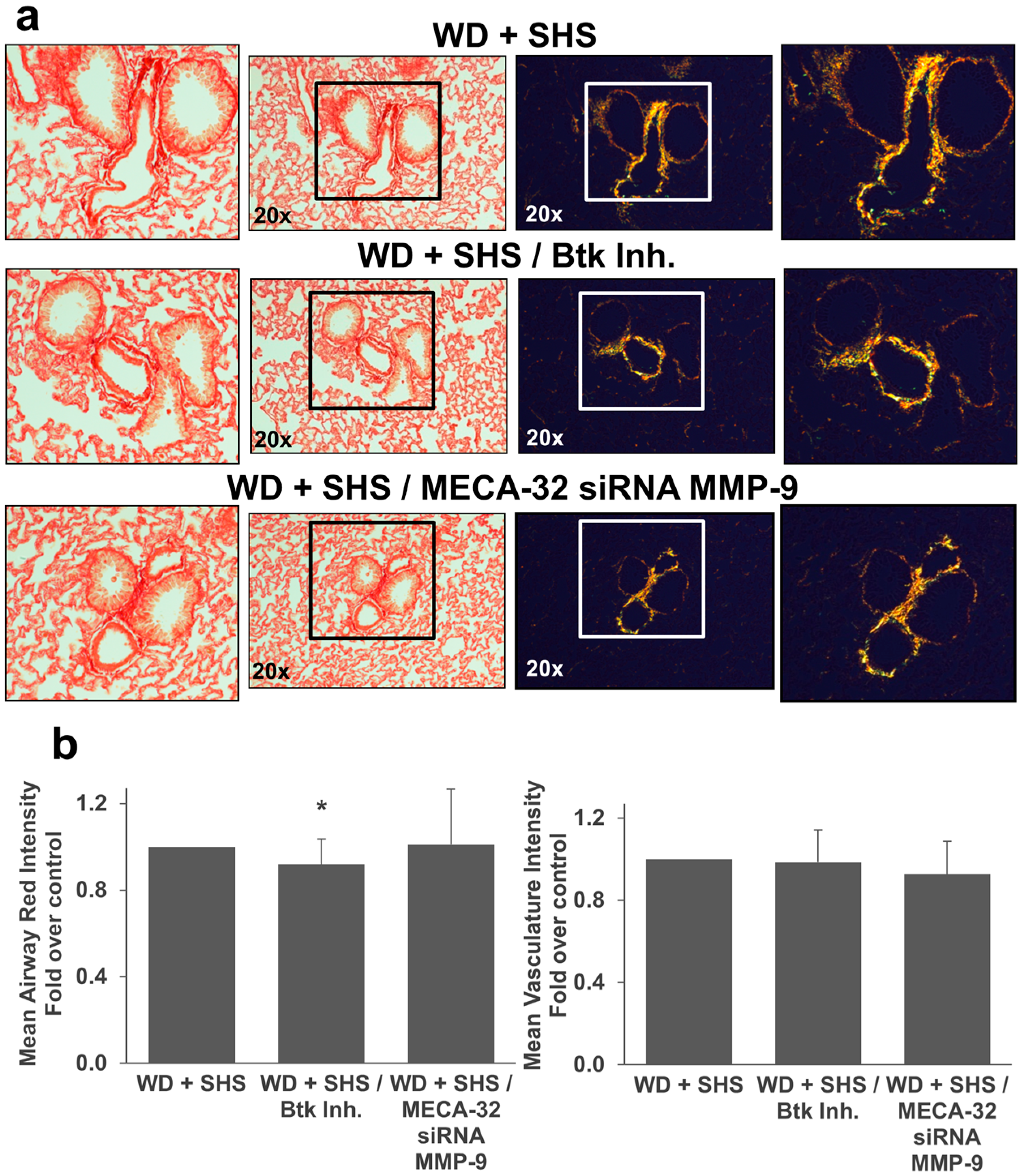
2.2. Exposure to SHS Triggers an Increase in Airway Wall Collagen in Lungs of ApoE−/− Mice
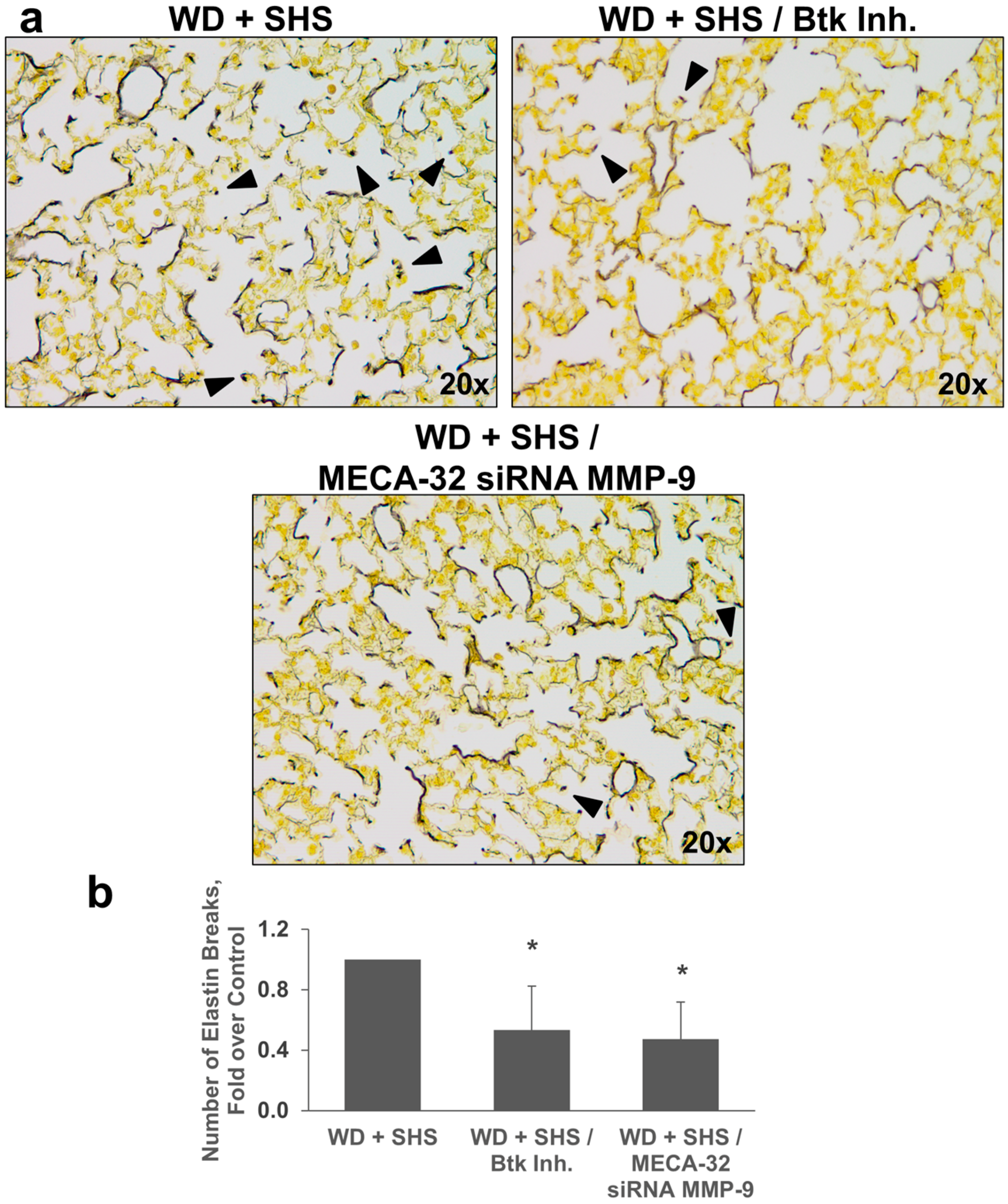
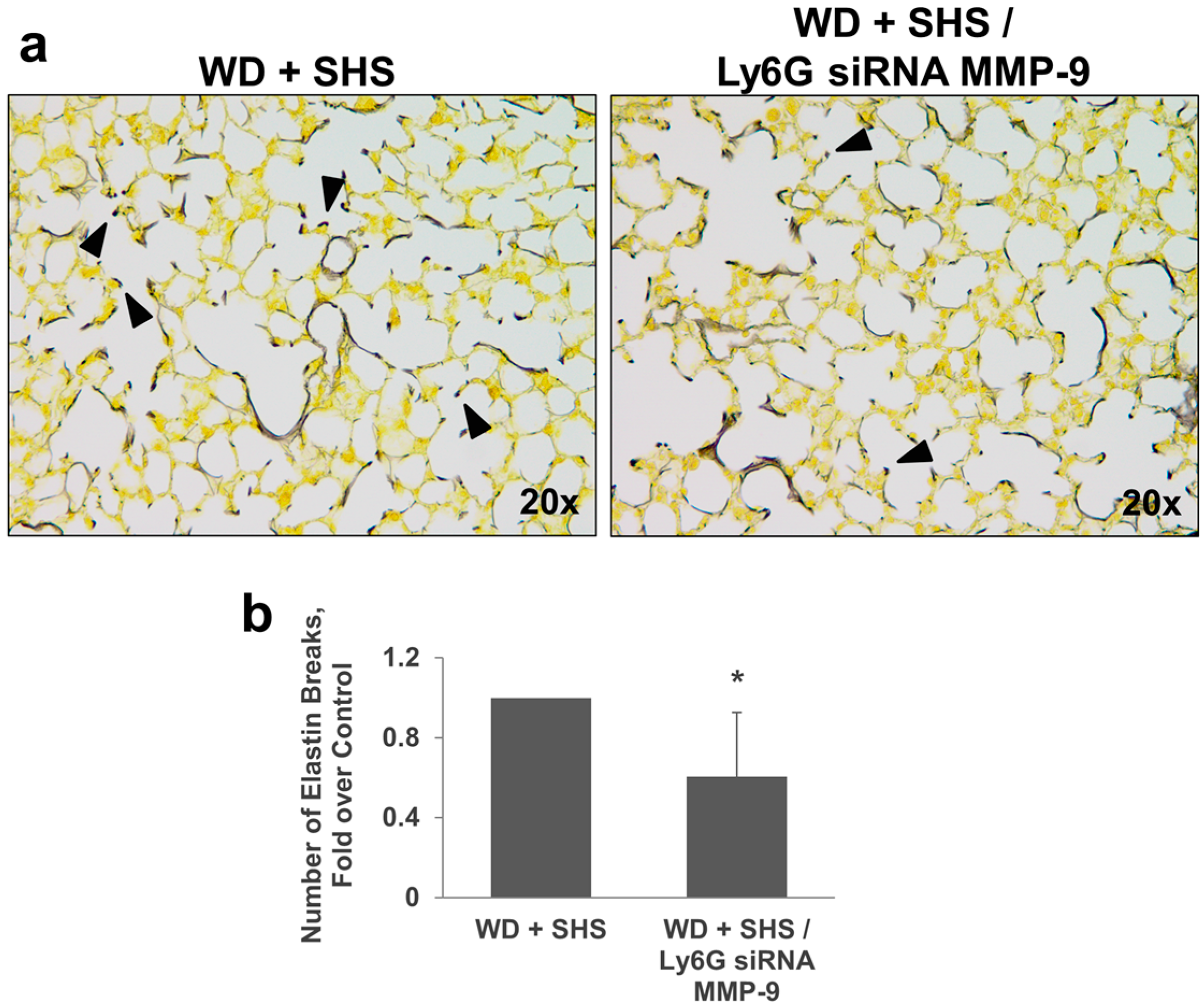
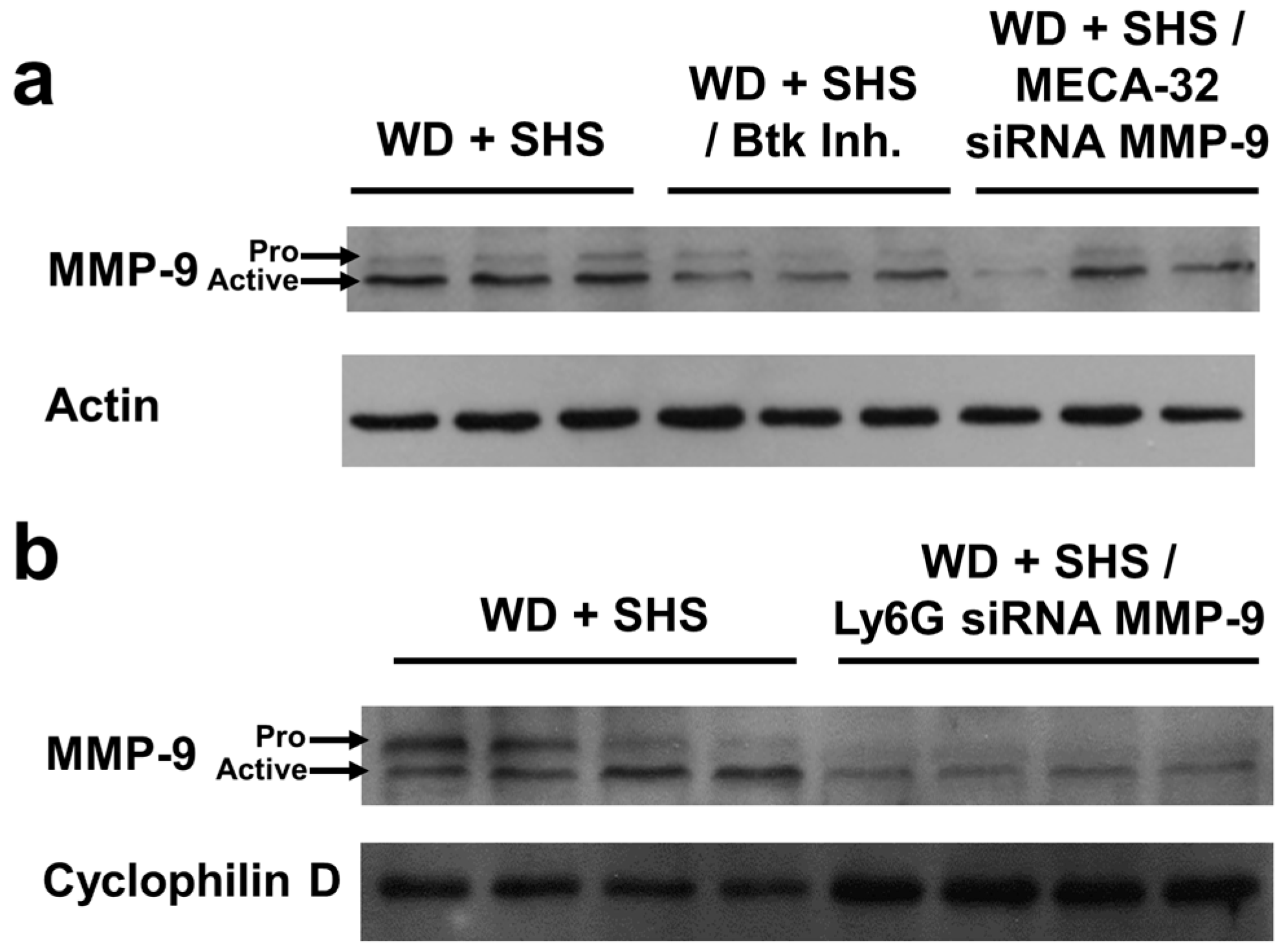
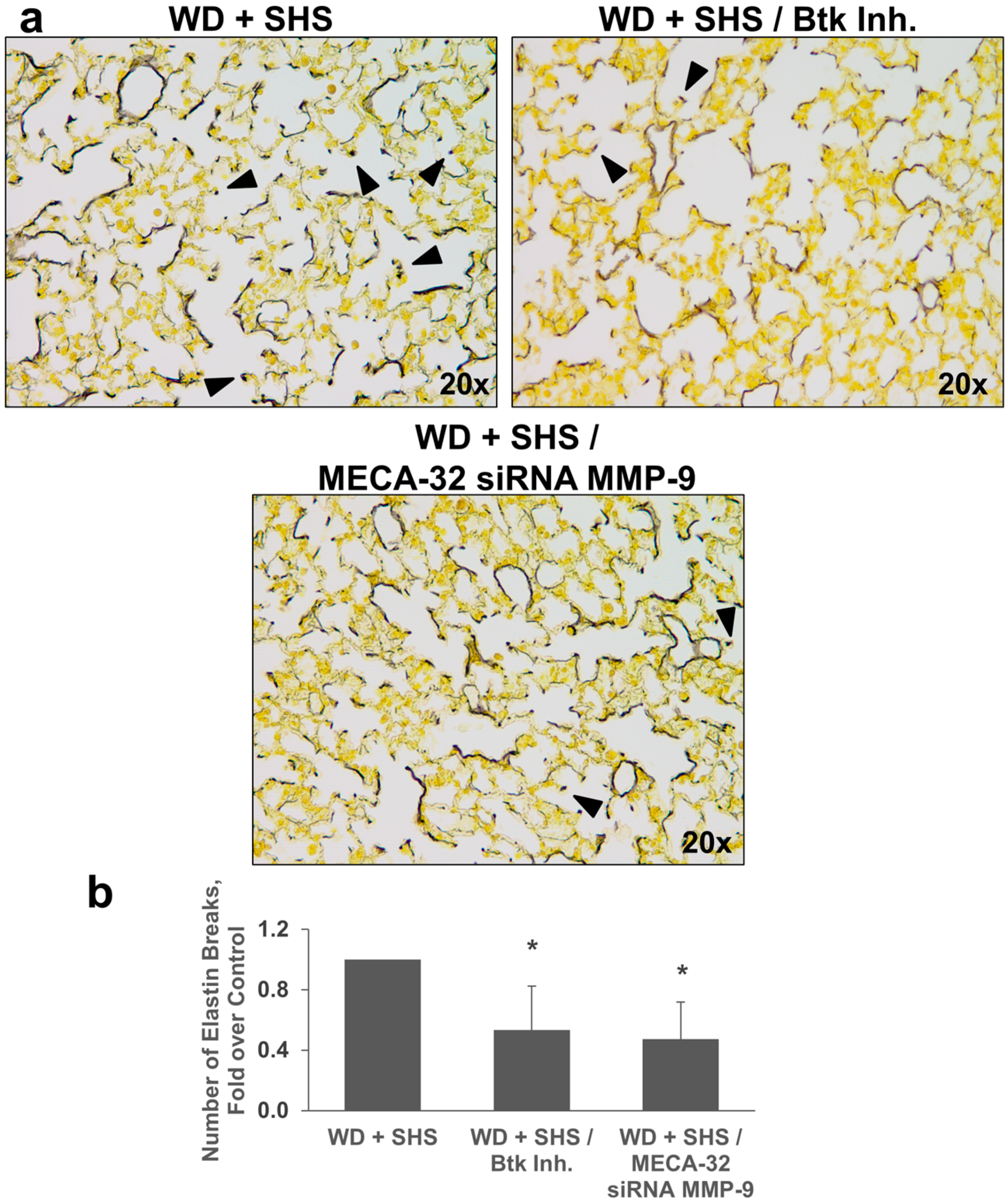
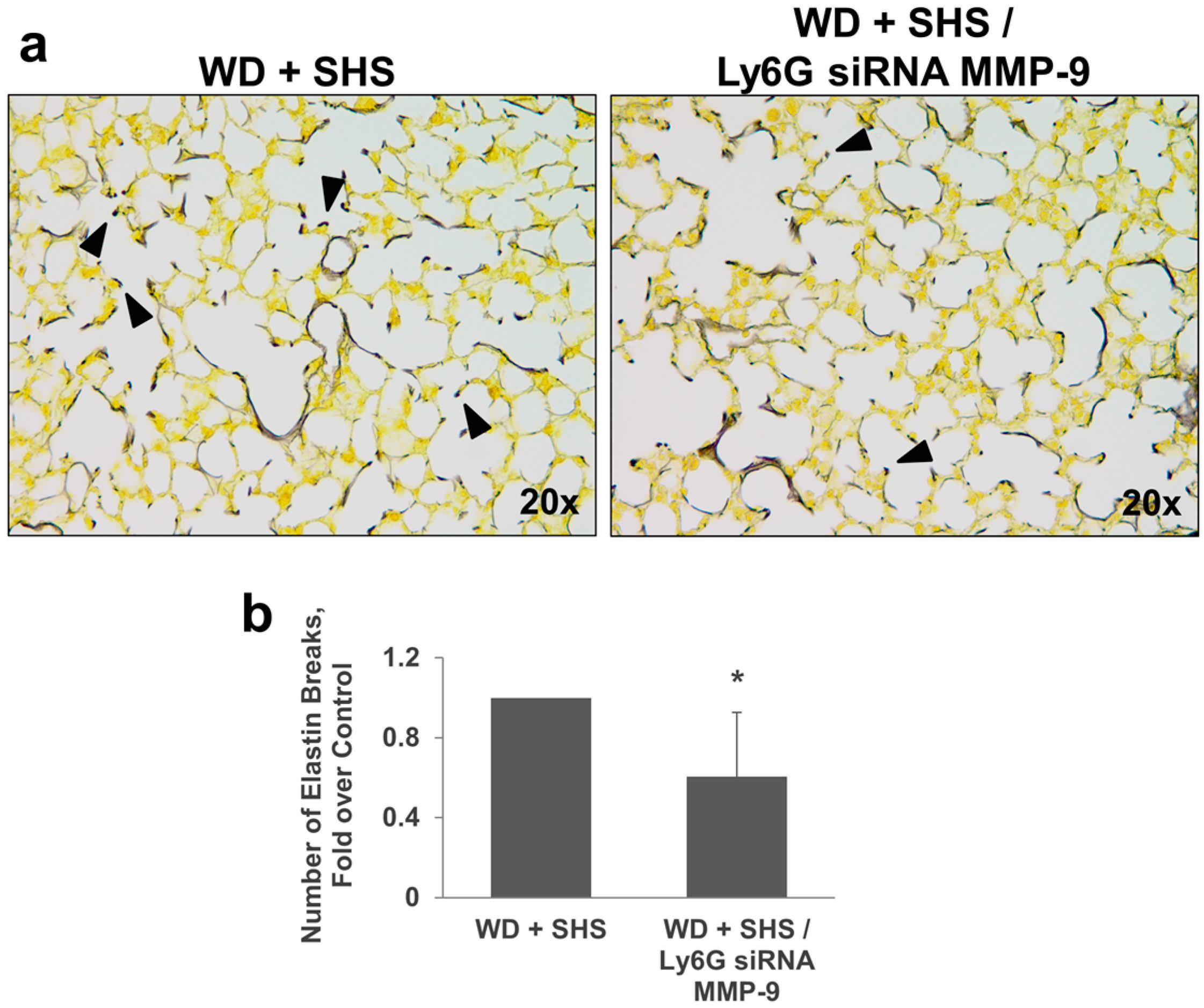
2.3. Treatments with a Pharmacological Inhibitor of Btk or MMP-9 Specific siRNA Targeting Either Endothelial Cells or Neutrophils Causes a Decrease in Alveolar Changes Related to COPD Progression
3. Discussion
4. Methods
4.1. Animal Studies
4.2. Histochemical Evaluation of Emphysemic Changes
4.3. Western Blotting
4.4. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ApoE−/− | apolipoprotein E-deficient |
| Btk | Bruton’s tyrosine kinase |
| Btk Inh. | Bruton’s tyrosine kinase inhibitor PCI-32765 (Ibrutinib) |
| COPD | Chronic obstructive pulmonary disease |
| Ly6G | Monoclonal antibody (clone 1A8) against Lymphocyte Antigen 6 Complex Locus G, a specific marker of mouse neutrophils |
| MECA-32 | Monoclonal antibody (clone MECA-32) against mouse pan-endothelial cell antigen, a specific marker of mouse endothelial cells |
| MMPs | matrix metalloproteinases |
| MMP-9 | matrix metalloproteinase-9 |
| PSR | picro-sirius red |
| SHS | second hand smoke |
| WD | Western Diet |
References
- Birru, R.L.; Di, Y.P. Pathogenic mechanism of second hand smoke induced inflammation and COPD. Front. Physiol. 2012, 3, 348. [Google Scholar] [CrossRef] [PubMed]
- Pappas, K.; Papaioannou, A.I.; Kostikas, K.; Tzanakis, N. The role of macrophages in obstructive airways disease: Chronic obstructive pulmonary disease and asthma. Cytokine 2013, 64, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 2014, 126, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Schlage, W.K.; Boué, S.; Veljkovic, E.; Peitsch, M.C.; Hoeng, J. The ApoE−/− mouse model: A suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J. Transl. Med. 2016, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, G.; Sundar, I.K.; Hwang, J.; Yao, H.; Rahman, I. Emphysema is associated with increased inflammation in lungs of atherosclerosis-prone mice by cigarette smoke: Implications in comorbidities of COPD. J. Inflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Boué, S.; Phillips, B.; Talikka, M.; Vihervaara, T.; Schneider, T.; Nury, C.; Elamin, A.; Guedj, E.; Peck, M.J.; et al. Effects of Cigarette Smoke, Cessation, and Switching to Two Heat-Not-Burn Tobacco Products on Lung Lipid Metabolism in C57BL/6 and ApoE−/− Mice—An Integrative Systems Toxicology Analysis. Toxicol. Sci. 2016, 149, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Naura, A.S.; Hans, C.P.; Zerfaoui, M.; Errami, Y.; Ju, J.; Kim, H.; Matrougui, K.; Kim, J.G.; Boulares, A.H. High-fat diet induces lung remodeling in ApoE-deficient mice: An association with an increase in circulatory and lung inflammatory factors. Lab. Investig. 2009, 89, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Beckett, E.L.; Stevens, R.L.; Jarnicki, A.G.; Kim, R.Y.; Hanish, I.; Hansbro, N.G.; Deane, A.; Keely, S.; Horvat, J.C.; Yang, M.; et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013, 131, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Jobse, B.N.; McCurry, C.A.; Morissette, M.C.; Rhem, R.G.; Stampfli, M.R.; Labiris, N.R. Impact of inflammation, emphysema, and smoking cessation on V/Q in mouse models of lung obstruction. Respir. Res. 2014, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Boue, S.; de Leon, H.; Schlage, W.K.; Peck, M.J.; Weiler, H.; Berges, A.; Vuillaume, G.; Martin, F.; Friedrichs, B.; Lebrun, S.; et al. Cigarette smoke induces molecular responses in respiratory tissues of ApoE−/− mice that are progressively deactivated upon cessation. Toxicology 2013, 314, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Fudala, R.; Florence, J.M.; Tucker, T.; Allen, T.C.; Standiford, T.J.; Luchowski, R.; Fol, M.; Rahman, M.; Gryczynski, Z.; et al. Bruton’s tyrosine kinase mediates cross-talk between FcγRIIa and TLR4 signaling cascades in human neutrophils. Am. J. Respir. Cell Mol. Biol. 2013, 48, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Fol, M.; Rahman, M.; Stokes, K.Y.; Florence, J.M.; Leskov, I.L.; Khoretonenko, M.V.; Matthay, M.A.; Liu, K.D.; Calfee, C.S.; et al. Silencing Bruton’s tyrosine kinase in alveolar neutrophils protects mice from LPS/immune complex induced acute lung injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L435–L448. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, C.A.; Doyle, S.; Brunner, C.; Dunne, A.; Brint, E.; Wietek, C.; Walch, E.; Wirth, T.; O’Neill, L.A. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor κB activation by Toll-like receptor 4. J. Biol. Chem. 2003, 278, 26258–26264. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Phillips, B.; Veljkovic, E.; Boue, S.; Schlage, W.K.; Vuillaume, G.; Martin, F.; Titz, B.; Leroy, P.; Buettner, A.; Elamin, A.; et al. An 8-Month Systems Toxicology Inhalation/Cessation Study in ApoE−/− Mice to Investigate Cardiovascular and Respiratory Exposure Effects of a Candidate Modified Risk Tobacco Product, THS 2.2, Compared With Conventional Cigarettes. Toxicol. Sci. 2016, 149, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef] [PubMed]
- Podowski, M.; Calvi, C.; Metzger, S.; Misono, K.; Poonyagariyagorn, H.; Lopez-Mercado, A.; Ku, T.; Lauer, T.; McGrath-Morrow, S.; Berger, A.; et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J. Clin. Investig. 2012, 122, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.M.; Tsai, L.W.; Ingenito, E.P.; Starcher, B.C.; Mariani, T.J. PPARgamma deficiency results in reduced lung elastic recoil and abnormalities in airspace distribution. Respir. Res. 2010, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucey, E.C.; Goldstein, R.H.; Stone, P.J.; Snider, G.L. Remodeling of alveolar walls after elastase treatment of hamsters. Results of elastin and collagen mRNA in situ hybridization. Am. J. Respir. Crit. Care Med. 1998, 158, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.V.; Abreu, S.C.; Padilha, G.A.; Rocha, N.N.; Maia, L.A.; Takiya, C.M.; Xisto, D.G.; Suki, B.; Silva, P.L.; Rocco, P.R.M. Characterization of a Mouse Model of Emphysema Induced by Multiple Instillations of Low-Dose Elastase. Front. Physiol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.; Weibel, E.; Gundersen, H.J.G.; Weinstein, F.V.; Ochs, M. Assessment of air space size characteristics by intercept (chord) measurement: An accurate and efficient stereological approach. J. Appl. Physiol. 2010, 108, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Postma, D.S.; Kerstjens, H.A.M.; Timens, W.; Whittaker, P.; Churg, A. Airway remodeling in the smoke exposed guinea pig model. Inhal. Toxicol. 2007, 19, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Brajer, B.; Batura-Gabryel, H.; Nowicka, A.; Kuznar-Kaminska, B.; Szczepanik, A. Concentration of matrix metalloproteinase-9 in serum of patients with chronic obstructive pulmonary disease and a degree of airway obstruction and disease progression. J. Physiol. Pharmacol. 2008, 59, 145–152. [Google Scholar] [PubMed]
- McAllister, D.A.; Maclay, J.D.; Mills, N.L.; Mair, G.; Miller, J.; Anderson, D.; Newby, D.E.; Murchison, J.T.; Macnee, W. Arterial Stiffness Is Independently Associated with Emphysema Severity in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 176, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Strulovici-Barel, Y.; Staudt, M.R.; Krause, A.; Gordon, C.; Tilley, A.E.; Harvey, B.G.; Kaner, R.J.; Hollmann, C.; Mezey, J.G.; Bitter, H.; et al. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax 2016, 71, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.; Peinado, V.I.; Ramírez, J.; Melgosa, T.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur. Respir. J. 2002, 19, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Can, U.; Yerlikaya, F.H.; Yosunkaya, S. Role of oxidative stress and serum lipid levels in stable chronic obstructive pulmonary disease. J. Chin. Med. Assoc. 2015, 78, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Austin, V.; Crack, P.J.; Bozinovski, S.; Miller, A.A.; Ross Vlahos, R. COPD and stroke: Are systemic inflammation and oxidative stress the missing links? Clin. Sci. 2016, 130, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Huang, Z.; Lin, H.; Ni, J.; Lu, H.; Chen, X.; Wang, Z.; Lin, L. Apolipoprotein E deficiency and high-fat diet cooperate to trigger lipidosis and inflammation in the lung via the Toll-like receptor 4 pathway. Mol. Med. Rep. 2015, 12, 2589–2597. [Google Scholar] [CrossRef] [PubMed]
- Khedoe, P.P.; Rensen, P.C.; Berbée, J.F.; Hiemstra, P.S. Murine models of cardiovascular comorbidity in chronic obstructive pulmonary disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L1011–L1027. [Google Scholar] [CrossRef] [PubMed]
- Meijer, M.; Rijkers, G.T.; Van Overveld, F.J. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Rev. Clin. Immunol. 2013, 9, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Rubinstein, I.; Kissel, T. siRNA delivery to the lung: What’s new? Adv. Drug Deliv. Rev. 2014, 75, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Alveolar macrophages as orchestrators of COPD. COPD 2004, 1, 59–70. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florence, J.M.; Krupa, A.; Booshehri, L.M.; Gajewski, A.L.; Kurdowska, A.K. Disrupting the Btk Pathway Suppresses COPD-Like Lung Alterations in Atherosclerosis Prone ApoE−/− Mice Following Regular Exposure to Cigarette Smoke. Int. J. Mol. Sci. 2018, 19, 343. https://doi.org/10.3390/ijms19020343
Florence JM, Krupa A, Booshehri LM, Gajewski AL, Kurdowska AK. Disrupting the Btk Pathway Suppresses COPD-Like Lung Alterations in Atherosclerosis Prone ApoE−/− Mice Following Regular Exposure to Cigarette Smoke. International Journal of Molecular Sciences. 2018; 19(2):343. https://doi.org/10.3390/ijms19020343
Chicago/Turabian StyleFlorence, Jon M., Agnieszka Krupa, Laela M. Booshehri, Adrian L. Gajewski, and Anna K. Kurdowska. 2018. "Disrupting the Btk Pathway Suppresses COPD-Like Lung Alterations in Atherosclerosis Prone ApoE−/− Mice Following Regular Exposure to Cigarette Smoke" International Journal of Molecular Sciences 19, no. 2: 343. https://doi.org/10.3390/ijms19020343