Genotyping by Sequencing Highlights a Polygenic Resistance to Ralstonia pseudosolanacearum in Eggplant (Solanum melongena L.)

 ,
,
Abstract
:
1. Introduction
- (i)
- Genotype a population of doubled haploid lines (DH) obtained from the hybrid [EG203 (encoded E4) × susceptible line MM 738 (encoded E8)] and construct a dense genetic map anchored on the tomato genome.
- (ii)
- (iii)
- Compare the position of EG203 resistance QTLs with those reported in other eggplant populations as well as in other solanaceous crops, thanks to the synteny of their genomes.
2. Results
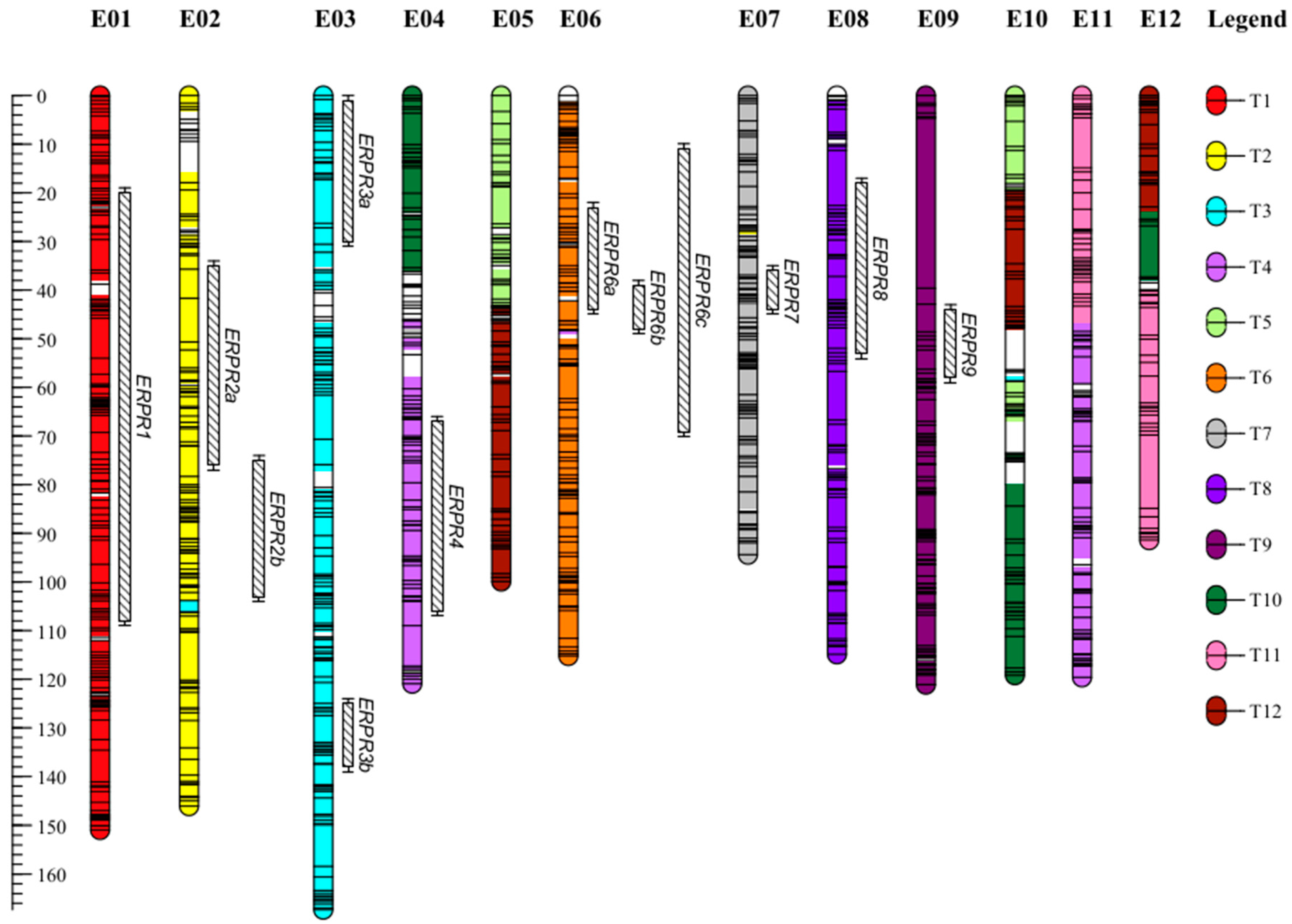
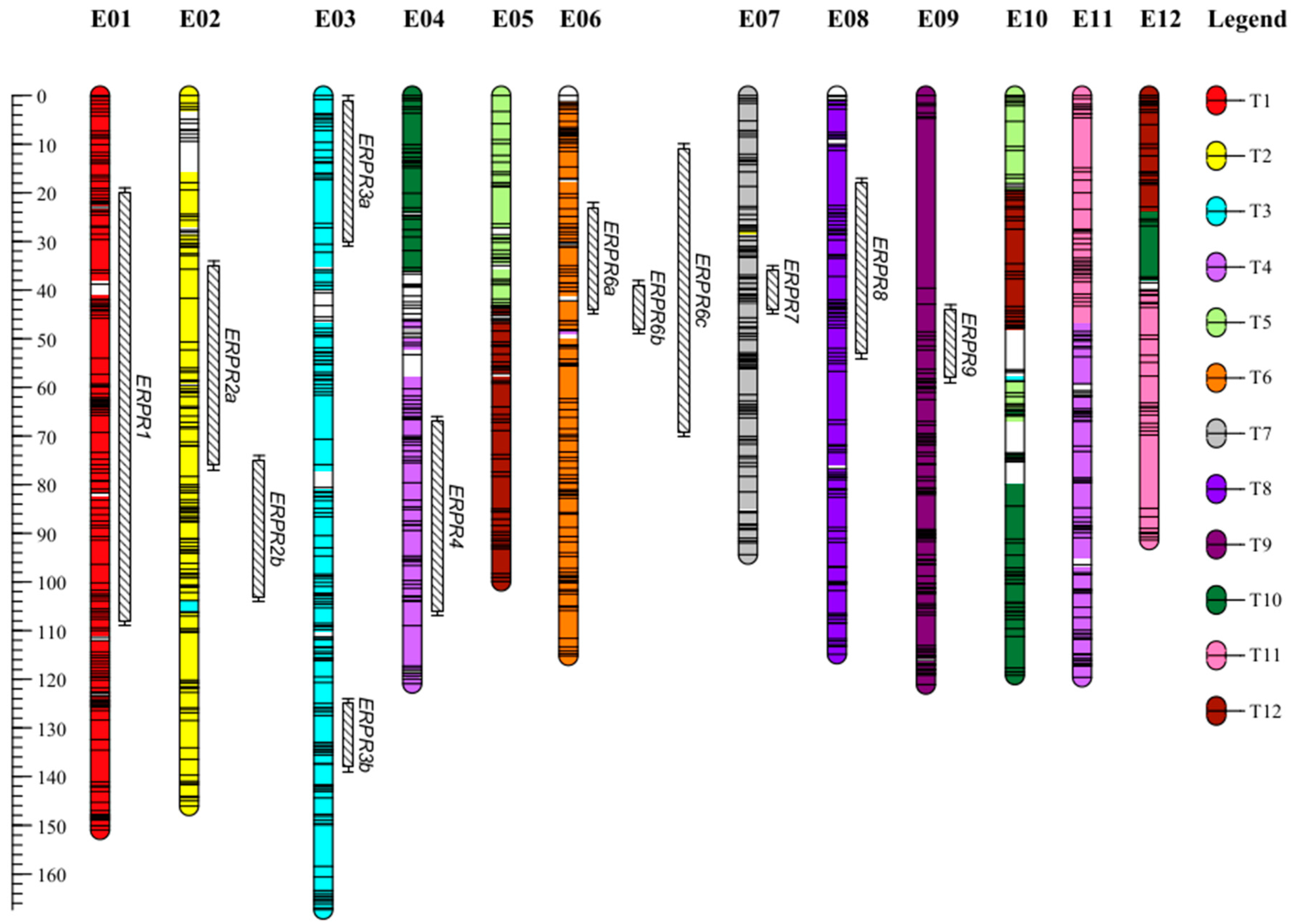
2.1. A Dense New Anchored Genetic Map for Eggplant
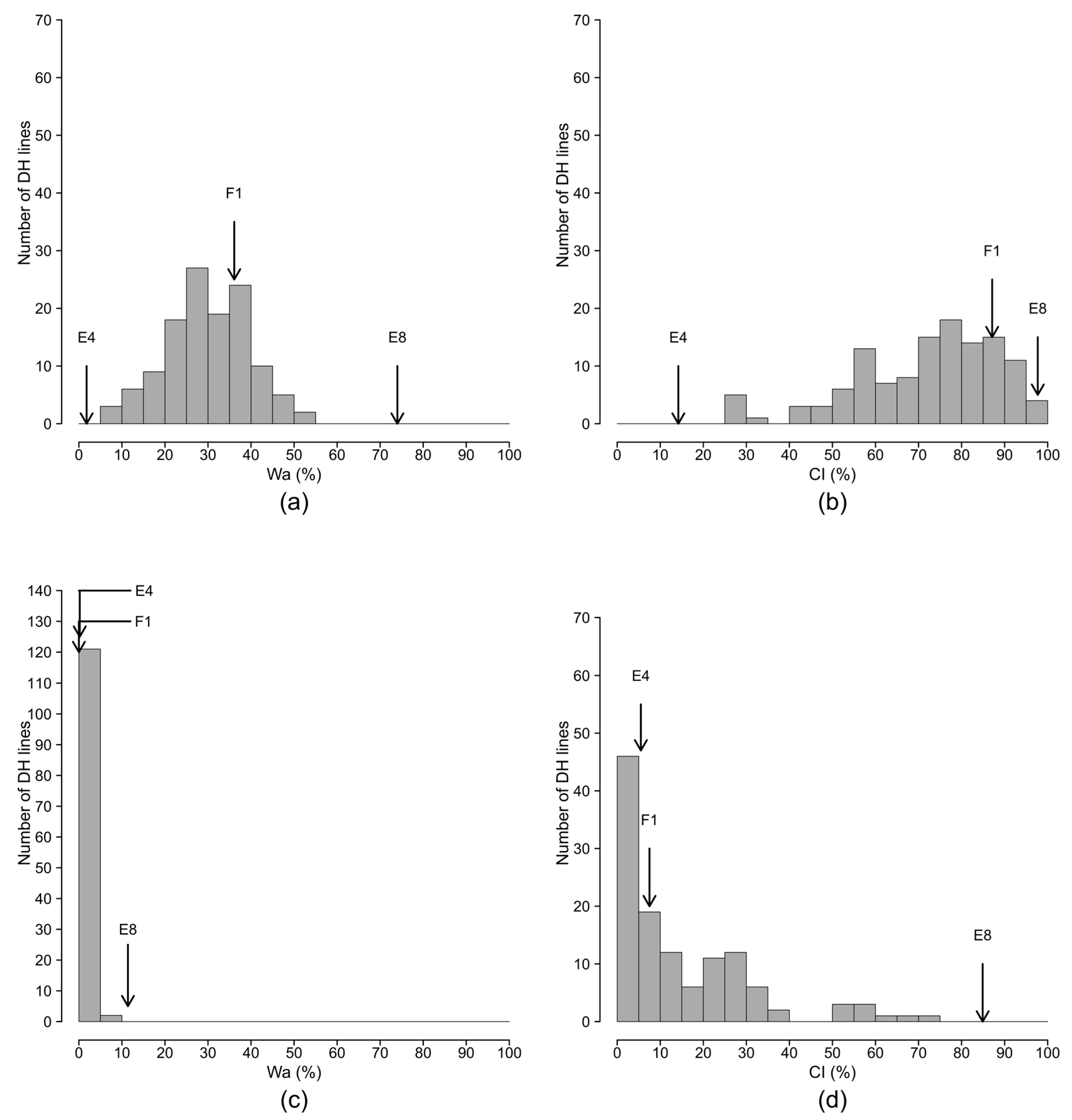
2.2. Segregation of Resistance in the Doubled Haploid Population
2.3. A Polygenic System Involved in the Resistance to PSS4 and R3598 Strains
2.4. Epistatic Effects Influence the Resistance to PSS4 Strain
- “More than additive” interactions (BB alleles of the resistant parent had a greater effect in the QTL duo combination, than individually) were observed for ERPR2b/ERPR6b and ERPR3a/ERPR6b pairs of loci.
- “Less than additive” interactions (BB alleles had a lower effect in combination than individually) were observed for ERPR2b/ERPR8, ERPR3a/ERPR8, and ERPR4/ERPR8 pairs of loci.
3. Discussion
3.1. SNPs from GBS Made It Possible to Construct a Dense New Intraspecific Genetic Map
3.2. Several QTLs Are Involved in the Resistance to R. pseudosolanacearum PSS4 and R3598 Strains and Are Possibly Syntenic with Proven Tomato BW-Resistance QTLs
3.3. Breeding Cultivars Resistant to R. pseudosolanacearum Strains Which Bypassed EBWR9
3.4. Looking for a Complementary Source of Resistance in Eggplant Germplasm
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. Library Construction and Sequencing
4.3. Sequence Analysis and Identification of SNPs
4.4. Genetic Map Construction
4.5. Bacterial Strains and Inoculum Preparation
4.6. Greenhouse Trials and Phenotyping
4.7. Statistical Analysis of Traits
4.8. QTL Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weese, T.L.; Bohs, L. Eggplant origins: Out of Africa, into the Orient. Taxon 2010, 59, 49–56. [Google Scholar]
- Daunay, M.-C. Eggplant. In Vegetables II: Fabaceae, Liliaceae, Solanaceae, and Umbelliferae; Prohens, J., Nuez, F., Eds.; Springer: New York, NY, USA, 2008; pp. 163–220. [Google Scholar]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant capacity of tea and common vegetables. J. Agric. Food Chem. 1996, 44, 3426–3431. [Google Scholar] [CrossRef]
- Niño-Medina, G.; Muy-Rangel, D.; Gardea-Béjar, A.; González-Aguilar, G.; Heredia, B.; Báez-Sañudo, M.; Siller-Cepeda, J.; Vélez de la Rocha, R.V. Nutritional and nutraceutical components of commercial eggplant types grown in Sinaloa, Mexico. Not. Bot. Horti Agrobot. Cluj-Napoca 2014, 42, 538–544. [Google Scholar] [CrossRef]
- Stommel, J.R.; Whitaker, B.D. Phenolic Acid Content and composition of eggplant fruit in a germplasm core subset. J. Am. Soc. Hortic. Sci. 2003, 128, 704–710. [Google Scholar]
- Plazas, M.; López-Gresa, M.P.; Vilanova, S.; Torres, C.; Hurtado, M.; Gramazio, P.; Andújar, I.; Herráiz, F.J.; Bellés, J.M.; Prohens, J. Diversity and relationships in key traits for functional and apparent quality in a collection of eggplant: Fruit phenolics content, antioxidant activity, polyphenol oxidase activity, and browning. J. Agric. Food Chem. 2013, 61, 8871–8879. [Google Scholar] [CrossRef] [PubMed]
- Prohens, J.; Rodríguez-Burruezo, A.; Raigón, M.D.; Nuez, F. Total phenolic concentration and browning susceptibility in a collection of different varietal types and hybrids of eggplant: Implications for breeding for higher nutritional quality and reduced browning. J. Am. Soc. Hortic. Sci. 2007, 132, 638–646. [Google Scholar]
- Peeters, N.; Guidot, A.; Vailleau, F.; Valls, M. Ralstonia solanacearum, a widespread bacterial plant pathogen in the post-genomic era. Mol. Plant Pathol. 2013, 14, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, A.C. Bacterial Wilt Caused By Pseudomonas solanacearum. J. Plant Pathol. 1991, 95, 237–245. [Google Scholar]
- Fegan, M.; Prior, P. How Complex Is the “Ralstonia solanacearum Species Complex”? Allen, C., Prior, P., Hayward, A.C., Eds.; APS Press: St. Paul, MN, USA, 2005; pp. 449–461. [Google Scholar]
- Safni, I.; Cleenwerck, I.; De Vos, P.; Fegan, M.; Sly, L.; Kappler, U. Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: Proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii s. Int. J. Syst. Evol. Microbiol. 2014, 64, 3087–3103. [Google Scholar] [CrossRef] [PubMed]
- Prior, P.; Ailloud, F.; Dalsing, B.L.; Remenant, B.; Sanchez, B.; Allen, C. Genomic and proteomic evidence supporting the division of the plant pathogen Ralstonia solanacearum into three species. BMC Genom. 2016, 17, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, T.A. Introduction and prospectus on the survival of R. solanacearum. In Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex; Allen, C., Prior, P., Hayward, A.C., Eds.; American Phytopathological Society (APS Press): Saint Paul, MN, USA, 2005; pp. 29–39. [Google Scholar]
- Genin, S.; Denny, T.P. Pathogenomics of the Ralstonia solanacearum species complex. Annu. Rev. Phytopathol. 2012, 50, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Saddler, G.S. Management of bacterial wilt disease. In Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex; Allen, C., Prior, P., Hayward, A.C., Eds.; American Phytopathological Society (APS Press): Saint Paul, MN, USA, 2005; pp. 121–132. [Google Scholar]
- Yamada, T.; Kawasaki, T.; Nagata, S.; Fujiwara, A.; Usami, S.; Fujie, M. New bacteriophages that infect the phytopathogen Ralstonia solanacearum. Microbiology 2007, 153, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Fujisawa, M.; Hamasaki, R.; Kawasaki, T.; Fujie, M.; Yamada, T. Biocontrol of Ralstonia solanacearum by treatment with lytic bacteriophages. Appl. Environ. Microbiol. 2011, 77, 4155–4162. [Google Scholar] [CrossRef] [PubMed]
- Addy, H.S.; Askora, A.; Kawasaki, T.; Fujie, M.; Yamada, T. Utilization of filamentous phage φRSM3 to control bacterial wilt caused by Ralstonia solanacearum. Plant Dis. 2012, 96, 1204–1209. [Google Scholar] [CrossRef]
- Boshou, L. A broad review and perspective on breeding for resistance to bacterial wilt. In Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex; Allen, C., Prior, P., Hayward, A.C., Eds.; APS Press: Saint Paul, MN, USA, 2005; pp. 225–238. [Google Scholar]
- Wang, J.F.; Ho, F.I.; Truong, H.T.H.; Huang, S.M.; Balatero, C.H.; Dittapongpitch, V.; Hidayati, N. Identification of major QTLs associated with stable resistance of tomato cultivar “Hawaii 7996” to Ralstonia solanacearum. Euphytica 2013, 190, 241–252. [Google Scholar] [CrossRef]
- Carmeille, A.; Caranta, C.; Dintinger, J.; Prior, P.; Luisetti, J.; Besse, P. Identification of QTLs for Ralstonia solanacearum race 3-phylotype II resistance in tomato. Theor. Appl. Genet. 2006, 113, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Mangin, B.; Thoquet, P.; Olivier, J.; Grimsley, N.H. Temporal and multiple quantitative trait loci analyses of resistance to bacterial wilt in tomato permit the resolution of linked loci. Genetics 1999, 151, 1165–1172. [Google Scholar] [PubMed]
- Thoquet, P.; Olivier, J.; Sperisen, C.; Rogowsky, P.; Laterrot, H.; Grimsley, N. Quantitative trait loci determining resistance to bacterial wilt in tomato cultivar Hawaii7996. Mol. Plant-Microbe Interact. 1996, 9, 826–836. [Google Scholar] [CrossRef]
- Thoquet, P.; Olivier, J.; Sperisen, C.; Rogowsky, P.; Prior, P.; Anais, G.; Mangin, B.; Bazin, B.; Nazer, R.; Grimsley, N. Polygenic resistance of tomato plants to bacterial wilt in the French West Indies. Mol. Plant-Microbe Interact. 1996, 9, 837–842. [Google Scholar] [CrossRef]
- Wang, J.F.; Olivier, J.; Thoquet, P.; Mangin, B.; Sauviac, L.; Grimsley, N.H. Resistance of tomato line Hawaii7996 to Ralstonia solanacearum Pss4 in Taiwan is controlled mainly by a major strain-specific locus. Mol. Plant Microbe Interact. 2000, 13, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, A.; Daunay, M.C.; Frary, A.; Palloix, A.; Wang, J.F.; Dintinger, J.; Chiroleu, F.; Wicker, E.; Prior, P. Bacterial Wilt Resistance in Tomato, Pepper, and Eggplant: Genetic Resources Respond to Diverse Strains in the Ralstonia solanacearum Species Complex. Phytopathology 2010, 101, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Mahbou-Somo-Toukam, G. Diversité de Ralstonia solanacearum au Cameroun et Bases Génétiques de la Résistance Chez le Piment (Capsicum Annuum) et Les Solanacées. Ph.D. Thesis, Institut des Sciences et Industries du Vivant et de l’Environnement (Agro Paris Tech), Paris, France, 2010. [Google Scholar]
- Mimura, Y.; Kageyama, T.; Minamiyama, Y.; Hirai, M. QTL analysis for resistance to Ralstonia solanacearum in Capsicum accession “LS2341”. J. Jpn. Soc. Hortic. Sci. 2009, 78, 307–313. [Google Scholar] [CrossRef]
- Mimura, Y.; Inoue, T.; Minamiyama, Y.; Kubo, N. An SSR-based genetic map of pepper (Capsicum annuum L.) serves as an anchor for the alignment of major pepper maps. Breed. Sci. 2012, 62, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ano, G.; Hebert, Y.; Prior, P.; Messiaen, C. A new source of resistance to bacterial wilt of eggplants obtained from a cross: Solanum aethiopicum L × Solanum melongena L. Agronomie 1991, 11, 555–560. [Google Scholar] [CrossRef]
- Lebeau, A.; Gouy, M.; Daunay, M.C.; Wicker, E.; Chiroleu, F.; Prior, P.; Frary, A.; Dintinger, J. Genetic mapping of a major dominant gene for resistance to Ralstonia solanacearum in eggplant. Theor. Appl. Genet. 2013, 126, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Salgon, S.; Jourda, C.; Sauvage, C.; Daunay, M.; Reynaud, B.; Wicker, E.; Dintinger, J. Eggplant resistance to the Ralstonia solanacearum species complex involves both broad-spectrum and strain-specific quantitative trait loci. Front. Plant Sci. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- N’Guessan, C.A.; Abo, K.; Fondio, L.; Chiroleu, F.; Lebeau, A.; Poussier, S.; Wicker, E.; Koné, D. So Near and Yet so Far: The Specific Case of Ralstonia solanacearum Populations from Côte d’Ivoire in Africa. Phytopathology 2012, 102, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.L.L.; Gardener, B.B.M.; Opina, N.; Miller, S.A. Diversity of Ralstonia solanacearum Infecting Eggplant in the Philippines. Phytopathology 2007, 97, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Pan, Z.C.; Prior, P.; Xu, J.S.; Zhang, Z.; Zhang, H.; Zhang, L.Q.; He, L.Y.; Feng, J. Genetic diversity of Ralstonia solanacearum strains from China. Eur. J. Plant Pathol. 2009, 125, 641–653. [Google Scholar] [CrossRef]
- Xue, Q.Y.; Yin, Y.N.; Yang, W.; Heuer, H.; Prior, P.; Guo, J.H.; Smalla, K. Genetic diversity of Ralstonia solanacearum strains from China assessed by PCR-based fingerprints to unravel host plant- and site-dependent distribution patterns. FEMS Microbiol. Ecol. 2011, 75, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Kim, J.; Kang, Y.; Lee, S.; Hwang, I. Genetic diversity and distribution of Korean isolates of Ralstonia solanacearum. Plant Dis. 2007, 91, 1277–1287. [Google Scholar] [CrossRef]
- Hirakawa, H.; Shirasawa, K.; Miyatake, K.; Nunome, T.; Negoro, S.; Ohyama, A.; Yamaguchi, H.; Sato, S.; Isobe, S.; Tabata, S.; et al. Draft genome sequence of eggplant (Solanum melongena L.): The representative solanum species indigenous to the old world. DNA Res. 2014, 21, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Fishman, L.; Kelly, A.J.; Morgan, E.; Willis, J.H. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 2001, 159, 1701–1716. [Google Scholar] [PubMed]
- Qiu, G.-F.; Xiong, L.-W.; Liu, Z.-Q.; Yan, Y.-L.; Shen, H. A first generation microsatellite-based linkage map of the Chinese mitten crab Eriocheir sinensis and its application in quantitative trait loci (QTL) detection. Aquaculture 2016, 451, 223–231. [Google Scholar] [CrossRef]
- Li, Y.; He, M. Genetic Mapping and QTL Analysis of Growth-Related Traits in Pinctada fucata Using Restriction-Site Associated DNA Sequencing. PLoS ONE 2014, 9, e111707. [Google Scholar] [CrossRef] [PubMed]
- Danesh, D.; Aarons, S.; McGill, G.E.; Young, N.D. Genetic dissection of oligogenic resistance to bacterial wilt in tomato. Mol. Plant-Microbe Interact. 1993, 7, 464–471. [Google Scholar] [CrossRef]
- Andolfo, G.; Sanseverino, W.; Rombauts, S.; Van de Peer, Y.; Bradeen, J.M.; Carputo, D.; Frusciante, L.; Ercolano, M.R. Overview of tomato (Solanum lycopersicum) candidate pathogen recognition genes reveals important Solanum R locus dynamics. New Phytol. 2013, 197, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Michelmore, R.W.; Meyers, B.C. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 1998, 8, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Mago, R.; Nair, S.; Mohan, M. Resistance gene analogues from rice: Cloning, sequencing and mapping. Theor. Appl. Genet. 1999, 99, 50–57. [Google Scholar] [CrossRef]
- Méndez-Vigo, B.; Rodríguez-Suárez, C.; Pañeda, A.; Ferreira, J.J.; Giraldez, R. Molecular markers and allelic relationships of anthracnose resistance gene cluster B4 in common bean. Euphytica 2005, 141, 237–245. [Google Scholar] [CrossRef]
- Young, R.A.; Melotto, M.; Nodari, R.O.; Kelly, J.D. Marker-assisted dissection of the oligogenic anthracnose resistance in the common bean cultivar,“G2333”. Theor. Appl. Genet. 1998, 96, 87–94. [Google Scholar] [CrossRef]
- Rodríguez-Suárez, C.; Méndez-Vigo, B.; Pañeda, A.; Ferreira, J.J.; Giraldez, R. A genetic linkage map of Phaseolus vulgaris L. and localization of genes for specific resistance to six races of anthracnose (Colletotrichum lindemuthianum). Theor. Appl. Genet. 2007, 114, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Geffroy, V.; Sicard, D.; de Oliveira, J.C.F.; Sévignac, M.; Cohen, S.; Gepts, P.; Neema, C.; Langin, T.; Dron, M. Identification of an ancestral resistance gene cluster involved in the coevolution process between Phaseolus vulgaris and its fungal pathogen Colletotrichum lindemuthianum. Mol. Plant-Microbe Interact. 1999, 12, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Gundry, C.N.; Vandersteen, J.G.; Reed, G.H.; Pryor, R.J.; Chen, J.; Wittwer, C.T. Amplicon melting analysis with labeled primers: A closed-tube method for differentiating homozygotes and heterozygotes. Clin. Chem. 2003, 49, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.B. Epistasis and plant breeding. Plant Breed. Rev. 2001, 21, 27–92. [Google Scholar]
- Causse, M.; Chaïb, J.; Lecomte, L.; Buret, M. Both additivity and epistasis control the genetic variation for fruit quality traits in tomato. Theor. Appl. Genet. 2007, 115, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Xi’ou, X.; Bihao, C.; Guannan, L.; Jianjun, L.; Qinghua, C.; Jin, J.; Yujing, C. Functional Characterization of a Putative Bacterial Wilt Resistance Gene (RE-bw) in Eggplant. Plant Mol. Biol. Rep. 2014, 33, 1058–1073. [Google Scholar] [CrossRef]
- Beavis, W.D. QTL analyses: Power, precision, and accuracy. In Molecular Dissection of Complex Traits; CRC Press: Boca Raton, FL, USA, 1998; pp. 145–162. [Google Scholar]
- Beavis, W.D. The power and deceit of QTL experiments: Lessons from comparative QTL studies. In Proceedings of the Forty-Ninth Annual Corn and Sorghum Industry Research Conference, Chicago, IL, USA, 5–7 December 1994; American Seed Trade Association: Chicago, IL, USA, 1994; pp. 250–266. [Google Scholar]
- Würschum, T.; Kraft, T. Cross-validation in association mapping and its relevance for the estimation of QTL parameters of complex traits. Heredity (Edinb) 2014, 112, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Göring, H.H.H.; Terwilliger, J.D.; Blangero, J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 2001, 69, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Utz, H.F.; Melchinger, A.E.; Schön, C.C. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 2000, 154, 1839–1849. [Google Scholar] [PubMed]
- Schön, C.C.; Utz, H.F.; Groh, S.; Truberg, B.; Openshaw, S.; Melchinger, A.E. Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 2004, 167, 485–498. [Google Scholar] [CrossRef] [PubMed]
- King, E.G.; Long, A.D. The Beavis effect in next-generation mapping panels in Drosophila melanogaster. G3 Genes Genomes Genet. 2017, 7, 1643–1652. [Google Scholar]
- Slate, J. From Beavis to beak color: A simulation study to examine how much QTL mapping can reveal about the genetic architecture of quantitative traits. Evolution 2013, 67, 1251–1262. [Google Scholar] [PubMed]
- Melchinger, A.E.; Utz, H.F.; Schön, C.C. Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 1998, 149, 383–403. [Google Scholar] [PubMed]
- Black, L.L.; Wu, D.L.; Wang, J.F.; Kalb, T.; Abbass, D.; Chen, J.H. Grafting Tomatoes for Production in the Hot-Wet Season; Asian Vegetable Research and Development Centre: Tainan, Taiwan, 2003; Volume 3, p. 551. [Google Scholar]
- Keatinge, J.D.H.; Lin, L.-J.; Ebert, A.W.; Chen, W.Y.; Hughes, J.D.A.; Luther, G.C.; Wang, J.-F.; Ravishankar, M. Overcoming biotic and abiotic stresses in the Solanaceae through grafting: Current status and future perspectives. Biol. Agric. Hortic. 2014, 30, 272–287. [Google Scholar] [CrossRef]
- Wicker, E.; Lefeuvre, P.; De Cambiaire, J.-C.; Lemaire, C.; Poussier, S.; Prior, P. Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 2012, 6, 961–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syfert, M.M.; Castañeda-álvarez, N.P.; Khoury, C.K.; Särkinen, T.; Sosa, C.C.; Achicanoy, H.A.; Bernau, V.; Prohens, J.; Daunay, M.C.; Knapp, S. Crop wild relatives of the brinjal eggplant (Solanum melongena): Poorly represented in genebanks and many species at risk of extinction 1. Am. J. Bot. 2016, 103, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.citeulike.org/user/nailest/article/11583827 (accessed on 7 September 2016).
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H.; De Koning, D.-J. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Stekhoven, D.J.; Bühlmann, P. MissForest—Non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. JoinMap® 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Plant Research International: Wageningen, The Netherlands, 2006; Volume 33, pp. 1310–1371. [Google Scholar]
- Chakravarti, A.; Lasher, L.K.; Reefer, J.E. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 1991, 128, 175–182. [Google Scholar] [PubMed]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [Green Version]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; NOVA GENETIC, Longué-Jumelles, France. Personal communication, 2015.
- Kelman, A. The relationship of pathogenicity of Pseudomonas solanacearum to colony appearance in a tetrazolium medium. Phytopathology 1954, 44, 693–695. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Italy, 2014; Available online: http://www.R-project.org/ (accessed on 1 July 2016).
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. lme4: Linear Mixed-Effects Models Using Eigen and S4. R Package Version. 2014. Available online: https://cran.r-project.org/web/packages/lme4/index.html (accessed on 21 October 2016).
- Hadfield, J.D. MCMC methods for multi-response generalized linear mixed models: The MCMCglmm R package. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Sen, Ś.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Botstein, S. Mapping factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [PubMed]
- Haley, C.S.; Knott, S.A. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 1992, 69, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [PubMed]
- Manichaikul, A.; Moon, J.Y.; Sen, Ś.; Yandell, B.S.; Broman, K.W. A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 2009, 181, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Hayter, A.J. The maximum familywise error rate of Fisher’s least significant difference test. J. Am. Stat. Assoc. 1986, 81, 1000–1004. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Strain | Season | Var a | Parents | Progenies of E8 × E4 | Control Lines | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E8 | E4 | F1 | F2 | BC1E8 | BC1E4 | DH | E3 | E6 | E9 | E10 | |||
| PSS4 | 1 | SCOmax | 3.7 (0.1) | 0.0 (0.0) | 2.2 (0.4) | 2.2 (0.1) | 3.6 (0.1) | 0.6 (0.0) | 1.8 (0.1) | 0.3 (0.0) | 2.2 (1.0) | 0.0 (0.0) | 3.7 (0.1) |
| Wa | 71.5 (2.1) | 0.4 (0.4) | 35.2 (4.8) | 38.1 (2.0) | 63.7 (1.2) | 12.7 (1.8) | 34.3 (1.0) | 7.4 (0.5) | 39.3 (18.9) | 0.0 (0.0) | 67.6 (1.7) | ||
| CI | 95.3 (2.7) | 4.1 (2.0) | 87.1 (7.1) | 75.6 (1.9) | 94.0 (3.0) | 38.2 (7.3) | 73.7 (1.2) | 15.4 (0.4) | 90.0 (0.0) | 17.2 (12.2) | 92.5 (2.5) | ||
| Pg | 5 | 1 | 4 | 4 | 5 | 2 | 4 | 2 | 4 | 1 | 5 | ||
| 2 | SCOmax | 4.0 (0.0) | 0.2 (0.1) | 2.3 (0.4) | 2.6 (0.2) | 3.5 (0.1) | 0.5 (0.2) | 2.7 (0.1) | 0.6 (0.4) | 3.4 (0.4) | 0.2 (0.2) | 3.3 (0.3) | |
| Wa | 76.6 (0.7) | 3.3 (1.2) | 37.0 (8.6) | 46.0 (5.3) | 62.2 (2.9) | 8.5 (2.0) | 41.4 (1.4) | 13.6 (9.6) | 60.3 (12.5) | 2.4 (2.4) | 57.8 (9.2) | ||
| CI | 100.0 (0.0) | 24.4 (8.2) | 87.1 (2.9) | 76.4 (6.3) | 93.4 (3.6) | 29.9 (9.7) | 82.0 (1.4) | 49.1 (14.1) | 95.0 (5.0) | 37.2 (7.2) | 89.4 (0.6) | ||
| Pg | 5 | 2 | 4 | 4 | 5 | 2 | 4 | 2 | 5 | 2 | 5 | ||
| Comb | SCOmax | 3.8 (0.1) | 0.1 (0.1) | 2.2 (0.2) | 2.4 (0.2) | 3.5 (0.1) | 0.6 (0.1) | 2.2 (0.1) | 0.5 (0.2) | 2.8 (0.6) | 0.1 (0.1) | 3.5 (0.2) | |
| Wa | 74.0 (1.7) | 1.8 (1.0) | 36.1 (4.1) | 42.0 (3.2) | 62.9 (1.4) | 10.6 (1.6) | 34.3 (1.0) | 10.5 (4.3) | 49.8 (11.1) | 1.2 (1.2) | 62.7 (4.7) | ||
| CI | 97.7 (1.7) | 14.2 (6.8) | 87.1 (3.1) | 76.0 (2.7) | 93.7 (1.9) | 34.0 (5.5) | 73.7 (1.2) | 32.2 (11.3) | 92.5 (2.5) | 27.2 (8.2) | 91.0 (1.4) | ||
| Pg | 5 | 1 | 4 | 4 | 5 | 2 | 4 | 2 | 4 | 2 | 5 | ||
| R3598 | 1 | SCOmax | 1.3 (0.6) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.6 (0.3) | 0.0 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.6 (0.6) |
| Wa | 11.4 (6.2) | 0.0 (0.0) | 0.0 (0.0) | 0.4 (0.4) | 4.9 (2.7) | 0.0 (0.0) | 0.5 (0.1) | 2.4 (2.4) | 0.2 (0.2) | 0.0 (0.0) | 3.9 (3.9) | ||
| CI | 77.8 (15.8) | 2.0 (2.0) | 5.0 (5.0) | 21.0 (3.0) | 61.0 (5.0) | 4.0 (1.0) | 16.3 (1.1) | 2.6 (2.6) | 40.0 (0.0) | 0.0 (0.0) | 74.7 (14.7) | ||
| Pg | 3.2 | 1 | 1 | 2 | 3.2 | 1 | 1 | 1 | 2 | 1 | 3.2 | ||
| 2 | SCOmax | 1.6 (0.4) | 0.1 (0) | 0.0 (0.0) | 0.4 (0.2) | 0.4 (0.1) | 0.0 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.0 (0.0) | 0.1 (0.1) | 0.4 (0.3) | |
| Wa | 11.5 (3.1) | 0.2 (0.2) | 0.0 (0.0) | 2.4 (1.2) | 2.5 (0.7) | 0.0 (0.0) | 0.4 (0.1) | 0.9 (0.9) | 0.1 (0.1) | 0.7 (0.7) | 3.3 (2.2) | ||
| CI | 92.0 (6.0) | 9.0 (3.0) | 10.0 (0.0) | 61.0 (8.0) | 81.0 (8.0) | 6.0 (1.0) | 12.1 (1.3) | 27.5 (7.5) | 20.0 (5.0) | 10.0 (0.0) | 72.5 (12.5) | ||
| Pg | 4 | 1 | 1 | 3.2 | 3.2 | 1 | 1 | 2 | 2 | 1 | 3.2 | ||
| Comb | SCOmax | 1.5 (0.3) | 0.0 (0.0) | 0.0 (0.0) | 0.2 (0.1) | 0.5 (0.2) | 0.0 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.5 (0.3) | |
| Wa | 11.4 (2.8) | 0.1 (0.1) | 0.0 (0.0) | 1.4 (0.8) | 3.7 (1.3) | 0.0 (0.0) | 0.5 (0.1) | 1.6 (1.1) | 0.2 (0.1) | 0.3 (0.3) | 3.6 (1.8) | ||
| CI | 84.9 (8.0) | 5.5 (2.5) | 7.5 (2.5) | 41.0 (12.1) | 71.0 (6.9) | 5.0 (0.8) | 16.3 (1.1) | 15.1 (7.9) | 30.0 (6.1) | 5.0 (2.9) | 73.6 (7.9) | ||
| Pg | 4 | 1 | 1 | 2 | 3.2 | 1 | 1 | 1 | 2 | 1 | 3.2 | ||
| Strain | Season | Var a | Sources of Variations b | h2 (Interval) c | |||
|---|---|---|---|---|---|---|---|
| G | S | R | G × S | ||||
| PSS4 | 1 | SCOmax | *** | - | ns | - | 0.68 (0.56–0.79) |
| Wa | *** | - | ns | - | 0.70 (0.59–0.80) | ||
| CI | *** | - | ** | - | 0.34 (0.23–0.45) | ||
| 2 | SCOmax | *** | - | ns | - | 0.57 (0.41–0.72) | |
| Wa | *** | - | ns | - | 0.58 (0.42–0.72) | ||
| CI | *** | - | ** | - | 0.36 (0.23–0.49) | ||
| Comb | SCOmax | *** | *** | ns | ** | 0.71 (0.59–0.80) | |
| Wa | *** | *** | ns | ** | 0.70 (0.57–0.80) | ||
| CI | *** | *** | *** | * | 0.50 (0.40–0.60) | ||
| R3598 | 1 | SCOmax | - | - | - | - | - |
| Wa | - | - | - | - | - | ||
| CI | *** | - | ns | - | 0.53 (0.36–0.68) | ||
| 2 | SCOmax | - | - | - | - | - | |
| Wa | - | - | - | - | - | ||
| CI | *** | - | ** | - | 0.56 (0.41–0.69) | ||
| Comb | SCOmax | - | - | - | - | - | |
| Wa | - | - | - | - | - | ||
| CI | *** | *** | * | ** | 0.39 (0.26–0.53) | ||
| Strains | Season | Var a | Chr b | QTL c | Pos d | Nearest Marker | Interval e | LOD | R2 f | Add Effect g | Total R2 h |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PSS4 | 1 | SCOmax | E03 | ERPR3a | 16.0 | s296164 | 3.0–29.0 | 3.8 | 13.2 | −0.38 *** | 13.2 |
| Wa | E03 | ERPR3a | 16.0 | s296164 | 2.0–27.2 | 3.4 | 11.9 | −9.11 *** | 11.9 | ||
| CI | E01 | ERPR1 | 76.8 | s219212 | 47.0–88.3 | 3.5 | 4.8 | −4.96 *** | 65.4 | ||
| E02 | ERPR2a | 40.0 | s135116 | 34.0–77.0 | 4.1 | 5.7 | −5.55 *** | ||||
| E03 | ERPR3a | 4.1 | s227496 | 0.0–13.0 | 7.3 | 10.8 | −7.69 *** | ||||
| E03 | ERPR3b | 126.7 | s231411 | 125.8–140.0 | 10.7 | 17.0 | −9.49 *** | ||||
| E04 | ERPR4 | 101.3 | s46892 | 99.0–102.0 | 8.5 | 13.0 | −7.23 *** | ||||
| E06 | ERPR6a | 23.0 | s902 | 22.0–33.0 | 10.2 | 16.0 | −7.32 *** | ||||
| E07 | ERPR7 | 41.6 | s272460 | 35.0–45.0 | 6.4 | 9.4 | −6.87 *** | ||||
| Epistasis | ERPR4:ERPR6a | 2.9 | 4.0 | −4.65 *** | |||||||
| 2 | SCOmax | E02 | ERPR2b | 98.4 | s51429 | 94.0–104.0 | 5.5 | 12.3 | −0.33 *** | 45.7 | |
| E04 | ERPR4 | 79.0 | s400 | 68.0–92.0 | 5.0 | 11.2 | −0.32 *** | ||||
| E06 | ERPR6b | 40.0 | s431 | 38.0–43.0 | 9.9 | 25.3 | −0.48 *** | ||||
| E08 | ERPR8 | 23.4 | s105566 | 18.0–28.6 | 5.0 | 11.5 | −0.31 *** | ||||
| Wa | E02 | ERPR2b | 98.4 | s51429 | 94.0–103.0 | 5.7 | 13.8 | −3.86 *** | 40.4 | ||
| E04 | ERPR4 | 88.4 | s208766 | 74.0–105.0 | 4.2 | 8.0 | −2.93 *** | ||||
| E06 | ERPR6b | 41.0 | s147256 | 38.6–49.0 | 7.5 | 16.8 | −4.41 *** | ||||
| E08 | ERPR8 | 23.4 | s105566 | 18.0–54.0 | 4.7 | 11.0 | −3.50 *** | ||||
| CI | E01 | ERPR1 | 38.8 | s231295 | 19.0–109.0 | 3.1 | 10.9 | −5.83 *** | 10.9 | ||
| Comb | SCOmax | E02 | ERPR2b | 98.4 | s51429 | 82.0–102.2 | 6.2 | 12.1 | −0.30 *** | 54.0 | |
| E03 | ERPR3a | 21.0 | s168838 | 2.0–31.0 | 3.5 | 6.4 | −0.23 *** | ||||
| E04 | ERPR4 | 71.3 | s53085 | 66.0–97.0 | 5.5 | 10.5 | −0.29 *** | ||||
| E06 | ERPR6b | 40.0 | s431 | 39.0–42.2 | 10.3 | 21.7 | −0.43 *** | ||||
| E08 | ERPR8 | 23.4 | s105566 | 18.0–28.6 | 5.5 | 10.4 | −0.28 *** | ||||
| Wa | E02 | ERPR2b | 98.4 | s51429 | 79.0–102.2 | 5.0 | 11.4 | −3.29 *** | 44.9 | ||
| E04 | ERPR4 | 97.0 | s311121 | 68.0–107.0 | 4.8 | 10.9 | −3.26 *** | ||||
| E06 | ERPR6b | 40.3 | s147256 | 39.0–43.0 | 7.7 | 18.5 | −4.28 *** | ||||
| E08 | ERPR8 | 23.4 | s105566 | 17.0–50.0 | 4.6 | 10.4 | −3.17 *** | ||||
| CI | E01 | ERPR1 | 68.0 | s168619 | 43.3–84.2 | 4.5 | 7.9 | −4.98 *** | 56.9 | ||
| E02 | ERPR2b | 87.7 | s131841 | 74.0–99.2 | 5.0 | 8.9 | −5.13 *** | ||||
| E03 | ERPR3a | 4.1 | s227496 | 2.0–27.0 | 4.5 | 7.8 | −4.98 *** | ||||
| E03 | ERPR3b | 127.5 | s231125 | 124.0–130.0 | 7.1 | 13.1 | −6.37 *** | ||||
| E04 | ERPR4 | 71.0 | s53085 | 66.6–79.6 | 6.6 | 12.0 | −6.22 *** | ||||
| E06 | ERPR6a | 39.4 | s56388 | 26.0–45.0 | 6.5 | 11.9 | −6.19 *** | ||||
| R3598 | 1 | SCOb | E03 | ERPR3a | 4.7 | s231544 | 0.0–28.0 | 3.2 | 11.4 | −1.09 *** | 11.4 |
| CI10b | E03 | ERPR3a | 5.0 | s197676 | 2.0–7.2 | 9.4 | 29.7 | −1.34 *** | 29.7 | ||
| CIb | E03 | ERPR3a | 5.0 | s197676 | 3.0–7.2 | 7.3 | 21.4 | −1.39 *** | 31.3 | ||
| E06 | ERPR6c | 46.0 | s681 | 39.0–65.0 | 4.3 | 12.0 | −1.06 *** | ||||
| 2 | SCOb | ns | ns | ns | ns | ns | ns | ns | ns | ns | |
| CI10b | E09 | ERPR9 | 51.5 | s75856 | 42.9–59.1 | 4.4 | 15.0 | −0.95 *** | 15.0 | ||
| CIb | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| Comb | SCO.b | ns | ns | ns | ns | ns | ns | ns | ns | ns | |
| CI10.b | E03 | ERPR3a | 4.1 | s227496 | 0.0–7.0 | 8.0 | 25.8 | −1.20 *** | 25.8 | ||
| CI.b | E03 | ERPR3a | 5.0 | s197676 | 0.0–14.0 | 4.0 | 12.6 | −1.04 *** | 21.6 | ||
| E06 | ERPR6c | 46.0 | s681 | 9.9–70.2 | 3.3 | 10.3 | −0.91 *** | ||||
| Pair of Loci | Genotypes at the First and Second Loci | |||
|---|---|---|---|---|
| AA/AA | AA/BB | BB/AA | BB/BB | |
| ERPR2b/ERPR6b | 2.72 ± 0.67 a (34) | 2.24 ± 0.83 a (32) | 2.31 ± 0.65 a (35) | 1.40 ± 0.83 b (22) |
| ERPR2b/ERPR8 | 2.72 ± 0.78 a (37) | 2.19 ± 0.69 b (29) | 2.09 ± 0.89 b (29) | 1.83 ± 0.79 b (28) |
| ERPR3a/ERPR6b | 2.60 ± 0.68 a (37) | 2.39 ± 0.96 a (21) | 2.41 ± 0.67 a (33) | 1.57 ± 0.76 b (32) |
| ERPR3a/ERPR8 | 2.82 ± 0.68 a (29) | 2.23 ± 0.80 b (29) | 2.15 ± 0.91 b (37) | 1.79 ± 0.65 b (28) |
| ERPR4/ERPR6b | 2.80 ± 0.65 a (26) | 2.22 ± 0.84 ab (32) | 2.34 ± 0.64 b (44) | 1.39 ± 0.83 c (21) |
| ERPR4/ERPR8 | 2.73 ± 0.78 a (33) | 2.15 ± 0.73 b (25) | 2.15 ± 0.88 b (33) | 1.90 ± 0.77 b (32) |
| Summary of QTLs Detected with PSS4 and R3598 Strains | BW-Resistance QTLs in Solanaceae Crops | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Interval (cM) | Position a | Strain | Name b | Species | Cultivar | Position a | Strains c | Studies d |
| ERPR2a | 34–77 | 37.6–43.9 | PSS4 | EBWR2 | Eggplant | AG91-25 | 38.3–46.9 | PSS4, TO10, CFPB2957, CFBP3059 | 8 |
| ERPR2b | 74–104 | 43.5–53.1 | PSS4 | ||||||
| ERPR3b | 124–139 | 65.2–67.8 | PSS4 | Bwr-3 | Tomato | Hawaii 7996 | 61.2–70.8 | GMI8217, JT516, Tm151 | 2,6,7 |
| ERPR6a | 22–45 | 0.0–37.8 | PSS4 | Bwr-6 | Tomato | Hawaii 7996, L285 | 36.9–38.8 | PSS4, PSS186, Tm151, JT519, UW364, JT516, GMI8217 | 1,2,3,4,5,6,7 |
| ERPR6b | 38–49 | 25.7–38.9 | PSS4 | ||||||
| ERPR6c | 39–65 | 25.7–41.9 | R3598 | ||||||
| Location | Season | Period of Assay | Strain | T (°C) | RH (%) |
|---|---|---|---|---|---|
| Reunion Island | 1 | November 2016 | PSS4 | 27 (±8) | 73 (±23) |
| 2 | May 2016 | PSS4 | 25 (±7) | 82 (±21) | |
| Cameroon | 1 | March 2016 | R3598 | 32 (±10) | 66 (±28) |
| 2 | June 2016 | R3598 | 28 (±7) | 74 (±23) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salgon, S.; Raynal, M.; Lebon, S.; Baptiste, J.-M.; Daunay, M.-C.; Dintinger, J.; Jourda, C. Genotyping by Sequencing Highlights a Polygenic Resistance to Ralstonia pseudosolanacearum in Eggplant (Solanum melongena L.). Int. J. Mol. Sci. 2018, 19, 357. https://doi.org/10.3390/ijms19020357
Salgon S, Raynal M, Lebon S, Baptiste J-M, Daunay M-C, Dintinger J, Jourda C. Genotyping by Sequencing Highlights a Polygenic Resistance to Ralstonia pseudosolanacearum in Eggplant (Solanum melongena L.). International Journal of Molecular Sciences. 2018; 19(2):357. https://doi.org/10.3390/ijms19020357
Chicago/Turabian StyleSalgon, Sylvia, Morgane Raynal, Sylvain Lebon, Jean-Michel Baptiste, Marie-Christine Daunay, Jacques Dintinger, and Cyril Jourda. 2018. "Genotyping by Sequencing Highlights a Polygenic Resistance to Ralstonia pseudosolanacearum in Eggplant (Solanum melongena L.)" International Journal of Molecular Sciences 19, no. 2: 357. https://doi.org/10.3390/ijms19020357
APA StyleSalgon, S., Raynal, M., Lebon, S., Baptiste, J.-M., Daunay, M.-C., Dintinger, J., & Jourda, C. (2018). Genotyping by Sequencing Highlights a Polygenic Resistance to Ralstonia pseudosolanacearum in Eggplant (Solanum melongena L.). International Journal of Molecular Sciences, 19(2), 357. https://doi.org/10.3390/ijms19020357