The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease


Department of Physiology and Cell Biology and The Zlotowski Center for Neuroscience, Faculty of Health Sciences, POB 653, Ben-Gurion Ave. Ben-Gurion University of the Negev, Beer Sheva 84105, Israel
Int. J. Mol. Sci. 2018, 19(2), 439; https://doi.org/10.3390/ijms19020439
Submission received: 7 January 2018
/
Revised: 24 January 2018
/
Accepted: 29 January 2018
/
Published: 1 February 2018
(This article belongs to the Special Issue Zinc Signaling in Physiology and Pathogenesis)
{kind=link}
Abstract
:A distinct G-protein coupled receptor that senses changes in extracellular Zn2+, ZnR/GPR39, was found in cells from tissues in which Zn2+ plays a physiological role. Most prominently, ZnR/GPR39 activity was described in prostate cancer, skin keratinocytes, and colon epithelial cells, where zinc is essential for cell growth, wound closure, and barrier formation. ZnR/GPR39 activity was also described in neurons that are postsynaptic to vesicular Zn2+ release. Activation of ZnR/GPR39 triggers Gαq-dependent signaling and subsequent cellular pathways associated with cell growth and survival. Furthermore, ZnR/GPR39 was shown to regulate the activity of ion transport mechanisms that are essential for the physiological function of epithelial and neuronal cells. Thus, ZnR/GPR39 provides a unique target for therapeutically modifying the actions of zinc in a specific and selective manner.
1. Introduction
The symptoms of zinc deficiency are particularly prominent in the digestive, immune, nervous, endocrine, and integumentary systems [1,2,3,4,5]. In many cases dietary zinc supplementation can ameliorate the symptoms and indeed zinc supplementation is widely used to treat diarrhea, the common cold, and skin conditions. The mechanisms underlying the roles of zinc have been revealed in the last two decades, but there is still a lot to learn about the pathways and regulation of zinc ions (Zn2+). Initially, Zn2+ was identified as a structural element and cofactor in enzymes [6,7] and transcription factors [8,9,10]. It is estimated that about 3000 proteins contain Zn2+ binding sites, and interaction with Zn2+ regulates or modulates the activity of these proteins, thereby affecting numerous cellular processes [11]. Cellular Zn2+ is associated with these proteins with a very high affinity and is considered a tightly bound pool of Zn2+ [10,12]. The labile Zn2+ pool in cells includes proteins that interact with Zn2+ via histidines, cysteines, or glutamate/aspartate residues; most prominent are the metallothioneiens (MTs) Zn2+ binding proteins [13]. This is a dynamic pool that releases Zn2+ upon redox signaling and oxidative or nitrosative stress, and contributes to cellular signaling [14,15,16,17,18]. In addition, cytosolic Zn2+ rise, likely mediated by Zn2+ transporters on the endoplasmic reticulum (ER), was monitored in mast cells following activation of the immunoglobulin receptor [19,20]. Subsequent studies determined that Zn2+ transporters found on various cellular organelles induce changes in cytosolic or organellar Zn2+ and thereby modulate cellular signaling [21,22,23,24,25,26]. Indeed, Zn2+ transport from the ER, Golgi, or mitochondria plays an important role in the function of mammary gland or prostate epithelial cells and other secretory cells [27,28,29]. Similar release of Zn2+, from the ER, during cardiac function regulates Ca2+ leakage from the ER in these cells [30,31]. These studies established Zn2+ as a second messenger that is released following diverse stimuli and triggers the regulation of kinases or phosphatases as well as protein expression [20,32]. Cellular Zn2+ is buffered by interaction with proteins and formation of complexes to rapidly reduce levels of Zn2+ to the picomolar range [17,33]. Importantly, transient changes in extracellular levels of Zn2+ can also occur following release of Zn2+-containing vesicles. Such vesicular Zn2+ is found in neurons, epithelial Paneth cells of the intestine or the salivary gland, as well as in pancreatic β-cells [34]. The vesicular Zn2+ can be released during normal activity of the cells; for example, Zn2+ is released into the synapse during neuronal activity or is secreted from β-cells or mammary epithelial cells [35,36,37,38,39,40]. Release of Zn2+ from cells can also occur following cellular injury and cell death, which liberates Zn2+ from the numerous Zn2+-binding proteins or cellular organelles [41]. Extracellular Zn2+ can interact with specific binding sites on numerous proteins and regulate their activity. For example, extracellular Zn2+ allosterically modulates numerous neuronal receptors, i.e., N-methyl-d-aspartate (NMDA), γ-Aminobutyric acid (GABA), or glycine receptors, thereby modulating the excitatory and inhibitory responses [42,43,44,45,46]. In epithelial cells, extracellular Zn2+ regulates the activity of purinergic receptors and the store-operated Ca2+ (SOC), representing an important link between Zn2+ and intracellular Ca2+ [47,48,49]. Application of Zn2+ was also suggested to upregulate the phosphatidylinositol-4,5-bisphosphate 3 (PI3) kinase/AKT pathway [50] or mitogen-activated protein kinases (MAPKs) [51], both essential to cell survival and proliferation.
2. Identification of a Zn2+-Sensing Receptor, ZnR/GPR39
In addition to the large numbers of Zn2+ homeostatic proteins described above, a distinct target for extracellular Zn2+ is the plasma membrane G-protein coupled receptor that is sensitive to Zn2+, ZnR/GPR39 [52,53,54]. G-protein coupled receptors are a large family of seven-transmembrane proteins that mediate cellular signaling in response to a diverse array of extracellular stimuli [55]. The endogenous Zn2+, released during physiological activity, acts as a first messenger and triggers intracellular Ca2+ signaling via the specific Gαq-coupled receptor ZnR/GPR39 [34,56]. Activity of ZnR/GPR39 in tissues relevant to Zn2+ signaling has been identified in neurons, colon epithelial cells (colonocytes), skin epidermal cells (keratinocytes), pancreatic cells, prostate cancer cells, salivary gland cells, and in bones [57,58,59,60,61]. In neurons, stimulation of the mossy fibers triggers ZnR/GPR39-dependent Ca2+ rises in postsynaptic CA3 (Cornu Ammonis 3) neurons [62] that are diminished in the presence of a non-permeable Zn2+ chelator, or in the absence of the Zn2+ transporter-3 (ZnT3), which is responsible for synaptic Zn2+ accumulation. Similar ZnR/GPR39 responses were observed in postsynaptic neurons of the auditory brainstem nucleus, the dorsal cochlear nucleus [63]. Importantly, ZnR/GPR39 activity was shown to enhance neuronal inhibitory tone, and zinc deficiency is associated with epilepsy and seizures, suggesting the significant physiological role of ZnR/GPR39 [53,64,65,66,67,68]. Luminal application of Zn2+ to colon epithelial cells, colonocytes, was sufficient to activate the plasma membrane ZnR/GPR39 [69], which is highly expressed in this tissue [70,71]. In colonocytes, ZnR/GPR39 activated cellular pathways that are strongly associated with cell growth, MAP, and PI3 kinases. The prominent role of zinc supplementation in digestive system function, taste disorders, and salivary secretion suggests that ZnR/GPR39 may play an important role in the physiological functions of this system. A specific role for zinc in wound healing and the strong link between its deficiency and skin lesions suggested that ZnR/GPR39 may mediate cell proliferation and wound healing, thereby contributing to skin health. A recent study also describes ZnR/GPR39 expression in the oviduct, where it colocalized with a higher concentration of Zn2+ but its activity has not been studied [72]. While a link to Zn2+ physiology is still not clear, ZnR/GPR39 was also associated with adipocyte and myoblast proliferation and differentiation [73,74]. Activation of ZnR/GPR39 was triggered by transient changes in extracellular Zn2+. While exogenous application of Zn2+ may trigger ZnR/GPR39 activation, the endogenous sources of vesicular Zn2+ may be the physiological trigger of ZnR/GPR39 activation, i.e., Zn2+ released from neuronal vesicles, salivary gland vesicles, pancreatic enzymes, or Paneth cells in the intestinal epithelium [35,36,37,38,39,40,75]. In addition, extracellular Zn2+ levels may transiently change following efflux mediated by Zn2+ transporters, such as ZnT6 [76], or following injury and cell death [41].
3. ZnR/GPR39-Dependent Signaling
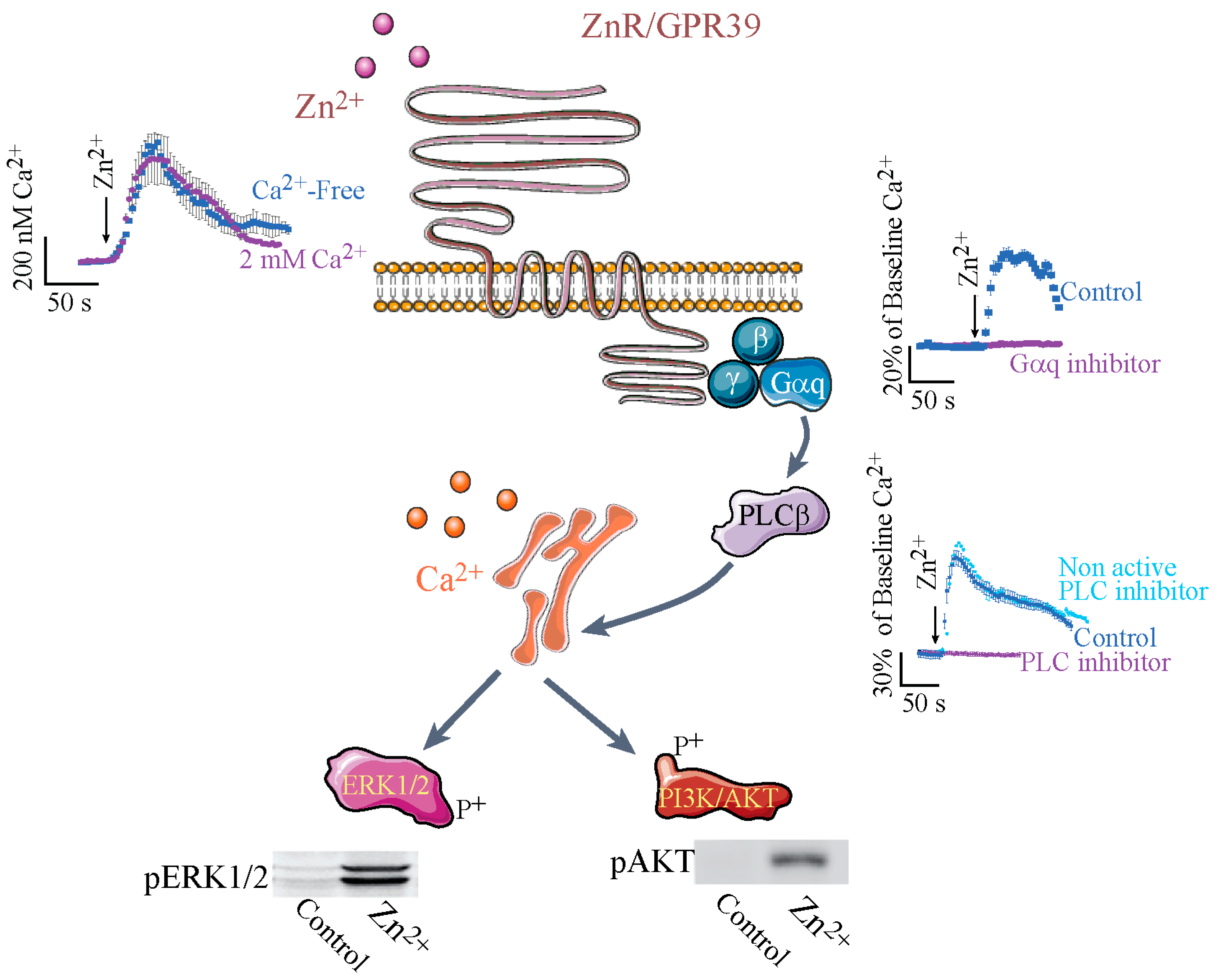
Intracellular Ca2+ signaling triggered by extracellular Zn2+ was the first functional identification of a distinct Zn2+ sensing receptor, named ZnR [77]. Use of pharmacological inhibitors of Gαq [78,79], inositol 1,4,5-trisphosphate (IP3) receptor and the phospholipase C (PLC), indicated that a Zn2+-dependent Ca2+ rise is mediated by activation of a Gαq-coupled receptor, such that the Ca2+ is released from thapsigargin-sensitive stores following activation of the IP3 receptor [52,57] (see Figure 1). Importantly, the Zn2+-dependent signaling was mediated by changes in extracellular, and not intracellular, levels of this ion, as expected from a G-coupled receptor [52,57]. The search for the protein that mediates Zn2+-dependent signaling focused on members of the Gαq family of receptors, their possible isoforms, or interactions between these receptors that may affect the affinity towards Zn2+; the main candidate in this family was the Ca2+-sensing receptor (CaSR). Most G-protein coupled receptors are activated by peptides and not cations, but a CaSR was already identified and its physiological significance to cellular signaling was established [80,81]. The similarity of the ligands and the signaling pathway activated by the CaSR and the putative ZnR suggested that these may be isoforms of the same receptor. Surprisingly, Zn2+ turned up in a screen of serum for the agonist of GPR39, which was an orphan receptor until then [82], subsequent studies confirmed that ZnR and GPR39 are one receptor, termed ZnR/GPR39. Despite their ligand similarity, CaSR and GPR39 are not members of the same subfamily of G-protein coupled receptors. The GPR39 is a member of ghrelin receptor family A, while CaSR is a member of family C of the G-protein coupled receptors [83]. It is important to note that ZnR/GPR39 is not activated by extracellular Ca2+, nor is the CaSR activated by Zn2+ [52,84]. Nevertheless, the affinity of ZnR/GPR39 to Zn2+ is modulated by Ca2+, as the K0.5 of ZnR/GPR39 in salivary gland cells was ~55 µM in the presence of Ca2+ and only ~36 µM in its absence [58]. This may be mediated by a direct effect of CaSR on ZnR/GPR39 conformation or its membrane expression or by a direct effect of Ca2+ on the Zn2+-binding site. Indeed, ZnR/GPR39 and the CaSR have been shown to directly interact in an exogenous overexpression system and may thereby modulate cation-dependent signaling in many systems where they are both expressed [84]. Importantly, the orphan receptor GPR39 was initially suggested to mediate signaling triggered by the obesity-related peptide obestatin [85]. These results were not reproduced by other laboratories and a study using serum identified Zn2+ as the endogenous ligand of GPR39 [82]. Using silencing and overexpression, it has been shown that the endogenous Zn2+-dependent signaling is mediated by GPR39, which is highly selective for Zn2+ and is not activated by Mn2+, Cu2+, or Fe2+ [52,53]. The affinity of ZnR/GPR39 to Zn2+ was physiologically adapted to the relevant tissues. For example, Zn2+ concentration in the digestive system lumen may reach hundreds of µM [86,87,88] and the colonocytic ZnR/GPR39 has an EC50 (half maximal effective concentration) of 80 µM [52,57]. Physiological relevance was further established when Zn2+ release from Caco-2 colonocytes was sufficient to induce ZnR/GPR39-dependent cell growth and tight junction formation [69,89]. In addition, in a cholera toxin model of diarrhea or a dextran sodium sulfate model of colitis, ZnR/GPR39-dependent pathways were not activated following dietary Zn2+ depletion [90,91]. Similarly, in the prostate, where there are high concentrations of Zn2+/citrate complex and transient release of this ion is likely to occur following cell death or changes in pH, ZnR/GPR39 is adapted to the relevant concentrations, which range from 10 to 200 μM [59]. In contrast, in keratinocytes ZnR/GPR39 EC50 to Zn2+ is in the nanomolar range, likely because this tissue contains much lower concentrations of labile Zn2+ [41]. Most importantly, the ZnR/GPR39 is triggered during keratinocytic injury, as shown using a scratch assay [41]. In addition, the neuronal ZnR/GPR39 has an affinity that is adapted to the release of Zn2+ from the synaptic mossy fiber terminals, and indeed very mild activation of these fibers induces sufficient Zn2+ levels to trigger postsynaptic ZnR/GPR39 signaling [62,92]. The differences in the affinity of the ZnR/GPR39 may result from its interaction with other, physiologically relevant G-protein coupled receptors in the tissues, as has been established for many receptors of this family [93].
Since Zn2+ can interact with numerous intracellular and extracellular proteins, application of this ion to study the effects of ZnR/GPR39 may yield confusing results and distinct agonists or antagonists would be of importance. Using various screening methods, agonists for ZnR/GPR39 have been suggested but very few were successfully tested in endogenous tissues. A recent study identified several compounds that may interact with ZnR/GPR39 and were shown to affect gastric function in wild-type but not GPR39 knockout mice, yet these compounds only potentiated the response of the ZnR/GPR39 to Zn2+ itself [94]. The use of molecular approaches to modulate expression of ZnR/GPR39, together with pharmacological inhibition of its signaling pathway, is therefore still important to study the effects of ZnR/GPR39. Indeed, the first description of the role of ZnR/GPR39 was established using a knockout mouse, which exhibited accelerated gastric emptying and increased body weight and fat composition [70]. This phenotype strengthened the link between the receptor and the well-known effects of Zn2+ on the gastrointestinal system. Future studies using knockout mice required challenging the mice to trigger a phenotypic distinction from the wild-type mice, suggesting that ZnR/GPR39 has a role in stress conditions. Finally, overexpression of ZnR/GPR39 in exogenous systems resulted in signaling that exhibited constitutive activity or was suggested to trigger Gαs or Gα12/13 signaling and CRE- or SRE-dependent gene expression [83], but the physiological significance of these pathways is yet to be determined.
Activation of the Gαq is triggering PLCβ activation and subsequent Ca2+ release from thapsigargin-sensitive ER stores. Insets show the Fura-2 fluorescent signals in cells expressing ZnR/GPR39 following application of Zn2+. The top left inset shows the calibrated level of Ca2+ change, monitored with Fura-2, obtained in the presence or absence of extracellular Ca2+; the right upper inset shows the % change of Ca2+ levels, relative to baseline Fura-2 fluorescence, in the presence or absence of the Gαq inhibitor (YM-254890); and the right bottom panel shows the % change of Ca2+ levels in the presence of the PLC inhibitor (U73122 active form, or U73343 inactive form). Subsequent to the Ca2+ signal ERK1/2 (extracellular regulated kinase) or AKT phosphorylation is monitored (shown in blots in the lower panels), indicating activation of the MAPK or PI3K pathways, respectively. (The figure was composed using Servier Medical Art templates (http://smart.servier.com/)).
Subsequent to the Ca2+ rise, ZnR/GPR39-triggers activation of the ERK/MAPK and AKT/PI3K pathways [57,84] that are essential for cell survival and proliferation [95]. ZnR/GPR39 activation in keratinocytes, colonocytes, and prostate cancer cells was shown to upregulate ERK and AKT phosphorylation and thereby cell growth. Activation of the Zn2+-dependent Ca2+ response was first shown to activate ERK1/2 phosphorylation, which was attenuated by functional de-sensitization of ZnR/GPR39, critical for protecting cells from excessive activation of the signaling [84]. In androgen-insensitive prostate cancer cell lines, ZnR/GPR39 activation by Zn2+ triggers PI3K pathway upregulation, which is reflected by increased expression and phosphorylation of AKT [84], associated with more malignant phenotypes of carcinomas [96,97,98]. Butyrate is a short-chain fatty acid found to affect colon epithelial cell growth and carcinogenesis [99,100,101,102]. In the colonocytic cell line, butyrate-induced apoptosis was attenuated by ZnR/GPR39-dependent activation of MAPK and PI3K pathways that increased expression of the pro-survival protein clusterin [69]. Moreover, enhanced cell proliferation was monitored using BrdU in colon tissue from ZnR/GPR39 expressing mice, but not in GPR39 knockout mice, during recovery from treatment with the toxin dextran sodium sulfate [90]. Under normal conditions BrdU staining in knockout mice lacking ZnR/GPR39 did not show differences from the wild-type tissue, suggesting that the baseline proliferation is intact, in agreement with the mild phenotype of these mice. The requirement for enhanced proliferation following the injury is the process that is impaired in the absence of ZnR/GPR39. As such, a role for ZnR/GPR39 may also underlie the healing effects of Zn2+ on gastric ulcers [103]. Topical application of zinc-containing ointments to enhance wound healing and re-epithelialization of the skin is well established [104,105,106,107]. Indeed ZnR/GPR39 activation in keratinocytes was shown to trigger MAPK phosphorylation and increased rate of scratch closure, suggesting that the receptor may mediate the effects of Zn2+ [41]. Finally, pre-adipocyte proliferation and differentiation are also induced following AKT activation, associated with ZnR/GPR39 expression [73,108]. In neurons, ZnR/GPR39 and subsequent Ca2+ release are essential for activation of MAPK by Zn2+ [92,109]. Such activation of the MAPK pathway by metabotropic signaling mediates changes in synaptic plasticity [110,111]. Finally, activation of ZnR/GPR39 in a salivary gland ductal cell line was shown to induce ATP release that mediated metabotropic signaling via the purinergic system in neighboring smooth muscle cells [58]. Thus ZnR/GPR39 has paracrine effects on neighboring cells, which may provide an important mechanism by which Zn2+ can affect physiological processes in tissues where not all cells express ZnR/GPR39 itself.
Zn2+, in contrast to most ligands of G-protein coupled receptors, is not rapidly degraded and a desensitization mechanism to protect cells from excessive Ca2+ signals is important for the regulation of ZnR/GPR39 signaling. Indeed, profound and prolonged desensitization [112] is monitored following exposure to subtoxic concentrations of Zn2+ [57,59,92]. The desensitization of ZnR/GPR39 by prolonged Zn2+ treatment induces internalization and possible degradation of the receptor, and profound loss of ZnR/GPR39 signaling is sustained for several hours. As such, Zn2+-induced desensitization was also used to specifically identify the roles of Zn2+ via ZnR/GPR39. For example, following ZnR/GPR39 desensitization the Zn2+-dependent ERK1/2 phosphorylation was diminished in prostate cancer cells [59]. The pathways that lead to ZnR/GPR39 desensitization are not fully understood. Recruitment of β-arrestin following ZnR/GPR39 with an allosteric modulator in the presence of Zn2+ did not induce desensitization but inhibition of Rho kinase blocked this process [113].
Zn2+ binding to ZnR/GPR39 occurs via two histidine residues, His17 and His19 [114], and an aspartate residue, Asp313. The pH sensitivity of these residues matched the regulation of ZnR/GPR39 response by extracellular pH. The ZnR/GPR39-dependent Ca2+ response and subsequent phosphorylation of MAP or PI3 kinase is completely abolished at pH 6.5 [41,109,115]. Hence, ZnR/GPR39 activity is regulated by physiologically relevant changes in extracellular Zn2+ or pH [115]. Thus, ZnR/GPR39 may be the mediator for many of the well-established, health-promoting functions of Zn2+ [116]. In contrast, local pH changes during inflammatory bowel disease may attenuate ZnR/GPR39-dependent cell proliferation in the digestive system and may contribute to epithelial erosion and barrier breakdown [117].
4. ZnR/GPR39 Regulation of Physiological Functions
4.1. ZnR/GPR39 Regulates Ion Transport Mechanisms
Downstream to activation of ZnR/GPR39, it has been shown that transport of Na+, K+, and Cl− are regulated. The movement of these ions is essential for the physiological functions of epithelial cells and neurons.
The ubiquitously expressed Na+/H+ exchanger (NHE) is upregulated following cytoplasmic acidification, to induce recovery of intracellular pH [118]. Activation of ZnR/GPR39 upregulates NHE activity in colonocytes, keratinocytes, and neurons [41,57,69,89,109], thereby providing a Zn2+-dependent homeostatic mechanism. Colonocytes are constantly exposed to cellular acidification, for example by short-chain fatty acid penetration [119], which can be recovered by NHE activity. Indeed, activation of ZnR/GPR39 in colonocytes and native colon tissues induced activation of NHE, downstream to the Ca2+ signaling and ERK1/2 activation, which enhanced the recovery of the colonocytic pH [57,69]. Thus, ZnR/GPR39 plays a role in pH homeostasis that is essential for colonocytes’ survival. Importantly, Na+-dependent H+ export can lower the extracellular pH. In keratinocytes, ZnR/GPR39 upregulation of NHE activity was also mediated via the same signaling pathway [41]. The extracellular acidification triggered by ZnR/GPR39-dependent activation of NHE may be required for migration of cells during wound healing or for the formation of an effective permeability barrier [120,121]. Intracellular acid loading in neurons affects neuronal excitability and results from metabolic H+ generation during repetitive firing [122]. Neuronal ZnR/GPR39 activation following release of Zn2+, concomitant with the neurotransmitter, resulted in increased NHE activity, thus relieving the metabolic acidification [109]. However, acidification of neuronal surfaces, by upregulating NHE activity, may contribute to tissue acidosis during ischemic neuronal injury [123]. Interestingly, ZnR/GPR39 itself is inactive at acidic pH [109], suggesting a homeostatic mechanism that can attenuate ZnR/GPR39 activation of NHE and excessive tissue acidification.
The K+/Cl− cotransporters (KCC) family is responsible for mediating Cl− efflux and thereby maintaining cell volume, as well as transepithelial ion transport and neuronal excitability [124]. These transporters are highly regulated via their phosphorylation and changes in surface expression [125,126]. In neurons, KCC2 is crucial for mediating Cl− efflux and thereby rendering the GABAA and glycine receptors inhibitory [127,128,129,130]. Activation of ZnR/GPR39 results in enhanced K+-dependent Cl− transport, which is mediated by KCC2 [62,131]. This Zn2+-dependent upregulation is abolished in the absence of ZnR/GPR39, or its downstream Ca2+ and MAPK activation. Moreover, Gαq-dependent signaling triggered by ZnR/GPR39 enhances KCC2 surface expression and thereby upregulates KCC2-dependent Cl− transport [62]. Similar upregulation of K+-dependent Cl− transport was also monitored following ZnR/GPR39 activation in colonocytes [91]. Loss of Cl− and Na+ into the colon lumen, via CFTR (cystic fibrosis transmembrane conductance regulator) upregulation for example, produces the driving force for water loss, thereby inducing diarrhea [132]. Yet, Cl− absorption pathways are not fully identified. Activation of ZnR/GPR39 in native colon epithelial tissue or in colonocytic cell lines resulted in activation of KCC1, which was mitogen activated kinase (MAPK)-dependent [91]. Moreover, KCC1 expression was shown to be basolateral, thereby providing a pathway for modulation of Cl− absorption in the colon.
4.2. ZnR/GPR39 Regulates Tight Junction Formation
Formation of epithelial barriers strongly depends on expression of junctional proteins, such as E-cadherin of the adherens junctions and zonula occludens-1 (ZO-1) or occludin of the tight junctions. This physical barrier is essential for the function of all epithelia and is particularly important in regions exposed to pathogens, such as the digestive tract. A role for Zn2+ in modulating colon epithelial tight junctions was previously described, but the prolonged application in that study may have resulted in changes of intracellular Zn2+ and not only activation of ZnR/GPR39 [133,134]. Using siRNA silencing of ZnR/GPR39 in Caco-2 colonocytic cell line revealed that ZnR/GPR39 was essential for Zn2+-dependent upregulation of tight junction formation, thus establishing that ZnR/GPR39 has a specific role in enhancing tight junctional complexes and epithelial barrier function [89]. It was further established that these colonocytic cells release Zn2+ in a manner that activates the ZnR/GPR39-dependent formation of the barrier, since a chelator of extracellular Zn2+ attenuated tight junction formation. Colon from ZnR/GPR39 deficient mice exhibited diminished expression level for the tight junction protein occludin, further revealing an important role of ZnR/GPR39 in barrier formation in vivo [90]. This loss of tight junctions may underlie some of the immune system effects associated with Zn2+ deficiency: as the permeation of pathogens is easier, inflammation may be prevalent during Zn2+ deficiency. A recent study showed that Zn2+ enhanced the expression of protein kinase C ζ (PKCζ), which was associated with ZnR/GPR39 levels, and linked to tight junction formation during Salmonella enterica serovar Typhimurium infection [135].
5. A Role for ZnR/GPR39 in Disease
5.1. ZnR/GPR39 in Wound Healing
Perhaps the oldest known use of zinc as a treatment is in dermal ointments for enhancing wound healing [104,105,106,107]. Zinc has been associated with proliferating tissues and is indeed accumulated in the skin [136]. Zinc transporters, i.e., ZIP4 (Zrt-Irt-like protein) or ZIP7, knockdown or mutations in these proteins also reveal an important role for these Zn2+ homeostatic proteins in skin formation during development [137]. Activation of ZnR/GPR39 in primary keratinocytes and in HaCaT cells suggested that Zn2+ may trigger this receptor signaling and may be the missing link between topical application of zinc and wound healing [41,52]. Indeed, the pro-proliferation/migration pathways were activated by ZnR/GPR39: ERK1/2 phosphorylation was increased via ZnR/GPR39-dependent activation of PKC and PI3K. In a scratch assay model, silencing of ZnR/GPR39 expression or activity inhibited the Zn2+-dependent increased rate of scratch closure [41]. One of the suggested benefits of zinc application was associated with anti-inflammatory effects. The ZnR/GPR39-dependent activation of NHE in keratinocytes induces acidification of the extracellular region. Such acidification is essential for reducing barrier permeability in the skin [120]. Hence NHE activation and the subsequent acidification by the ZnR/GPR39 may also exert an anti-inflammatory effect. Finally, if paracrine release of ATP following ZnR/GPR39 activation [58] also occurs in keratinocytes, it suggests another mechanism to increase the proliferation of neighboring fibroblasts that do not express ZnR/GPR39. Activation of cellular signaling by ZnR/GPR39 may affect numerous pathways and Zn2+ binding proteins. As such, a role for MG53, a Zn2+ binding protein, has been associated with myoblasts’ cell membrane recovery following permeation of Zn2+ into the cells [138]. The ZnR/GPR39 has also been described in myogenic processes, but the role of Zn2+ in this aspect has not been addressed [74], hence future studies on the role of ZnR/GPR39 in muscle cell recovery would be of interest.
5.2. Diarrhea and Inflammatory Bowel Diseases
Prominent roles of Zn2+ include attenuation of diarrhea and amelioration of symptoms of inflammatory ulcerative disease, such as Crohn’s disease and colitis [139,140,141,142,143]. Initial breakdown of tight junctions is considered a trigger to recurrence of inflammatory bowel diseases. The ZnR/GPR39-dependent enhancement of junctional complex proteins ZO-1 and occludin [69,89] suggested that ZnR/GPR39 may be involved in ameliorating symptoms of inflammatory bowel diseases. Indeed, ZnR/GPR39 deficient mice showed increased susceptibility to dextran sodium sulfate (DSS) model of colitis [90]. Even more profound was the effect of ZnR/GPR39 during a recovery phase. ZnR/GPR39 expression was essential for rapid recovery of the epithelial layer, via increased proliferation and crypt formation, and formation of the physical barrier, via increased expression of occludin. The benefit of Zn2+ treatment in inflammatory bowel disease is controversial. The results described here suggest that during bouts of the inflammatory state the epithelial erosion and loss of ZnR/GPR39 on the epithelial barrier may render Zn2+ inefficient, yet if provided during remission Zn2+, via ZnR/GPR39, may extend this period. In fact, ZnR/GPR39 expression in the epithelial cells may serve as a therapeutic target that can be specifically activated to extend the remission periods.
Maintenance of osmotic gradients, for proper water movement, is mediated by ion transporters found on the epithelial cells [144]. In diarrhea, impaired transporters function results in excessive loss of Na+ and Cl− into the lumen and subsequent water loss. The Zn2+ and ZnR/GPR39 upregulation of Na+/H+ exchanger activity [57,69] can serve to enhance uptake of Na+ from the lumen. Indeed many previous studies showed that the colonocytic apical NHE3 upregulation enhances Na+ absorption and thereby reduces water loss and diarrhea [144,145,146]. In addition, activation of a basolateral KCC1 by ZnR/GPR39 increases absorption of Cl−, which is also essential to reducing fluid loss. Cholera toxin infection, a common cause of diarrhea, induced significantly worse diarrhea in GPR39 knockout mice, lacking ZnR/GPR39 signaling, compared to WT mice [91]. Thus, ZnR/GPR39 activation can reduce fluid loss during the disease, but reduced luminal Zn2+, which may be a dietary or disease-mediated condition, may diminish the protective effect of this pathway. While Zn2+ is suggested by the World Health Organization (WHO) as an important supplement to treat diarrhea [142,147], ZnR/GPR39 is a novel and specific target that may be more effectively targeted.
5.3. Epilepsy
Several studies linked the loss of synaptic Zn2+ or Zn2+ deficiency with increased incidence of seizures [148,149,150,151]. Despite a well-known role for Zn2+ in modulating numerous excitatory and inhibitory post synaptic targets, how synaptically released Zn2+ can affect epileptogenesis was not clear. Nevertheless, the major phenotype of the ZnT3 knockout mice, lacking synaptic Zn2+, is enhanced sensitivity to kainate-induced or febrile hyperthermia induced seizures [152,153]. This indicated that synaptic Zn2+ itself does have a role in epilepsy. The results showing regulation of Cl− transport by ZnR/GPR39 activation of KCC2, taken together with the prominent role of loss of KCC2 function in increasing seizure susceptibility [128,154,155], suggested that ZnR/GPR39 may play a role in epilepsy via this pathway. Indeed, GPR39 knockout animals, lacking ZnR/GPR39 signaling, exhibit enhanced susceptibility to kainate-induced seizures, with significantly higher behavioral seizure severity scores and more seizures over longer periods of time compared to wild-type controls [156]. Kainate-induced upregulation of KCC2 activity is dependent on Zn2+, which is released by the increased firing under these enhanced excitability conditions. Moreover, ZnR/GPR39 signaling via the Gαq and subsequent MAPK pathway are required for increased KCC2 activity and thereby inhibitory tone. Thus the homeostatic role of ZnR/GPR39, activated by Zn2+ co-released with glutamate, is essential during excessive firing to reduce excitatory activity via enhancing GABAergic responses. In contrast, loss of this signaling in the absence of synaptic Zn2+ or ZnR/GPR39 may result in epileptogenesis [53]. A similar effect on increasing inhibitory neuronal signaling is monitored in the dorsal cochlear neurons, where ZnR/GRP39 activation enhances endocannabinoid release and reduces excitatory glutamate release [63]. In addition, enhanced excitability and thereby seizure activity has been associated with neuronal acidification and loss of Na+/H+ exchanger (NHE) activity [157,158]. Thus ZnR/GPR39-dependent upregulation of NHE activity, which was monitored in primary neurons, may also link the receptor to reduced seizures [109]. In Alzheimer’s disease, Aβ oligomers interact with Zn2+ [159,160], thus lowering levels of labile Zn2+. Indeed, in the presence of Aβ, the ZnR/GPR39-dependent Ca2+ responses in primary neurons were significantly reduced and resulted in much lower MAPK activation [161]. This decrease in ZnR/GPR39-dependent signaling, reducing the homeostatic activation of KCC2, may serve as a link to the increased incidence of seizure found in Alzheimer’s disease patients compared to the general population.
5.4. Depression
Zinc deficiency is associated with neurological and psychiatric disorders [162]; however, it is not yet clear if the decrease in Zn2+ results from aberrant intake, especially in depression, when appetite is lost and general uptake of nutrients is low, or is a cause of the disorder. Several studies reported a role for ZnR/GPR39 in depression, based on apparent changes in the expression level of this receptor following Zn2+-deficiency that were correlated with behavioral changes, also in suicide victims [163,164]. Changes in ZnR/GPR39 expression were also shown following treatment with monoaminergic inhibitors, such as used to treat depression, thus suggesting a link between the receptor and this disease. Surprisingly, despite the extensive use of antibodies against ZnR/GPR39 in this study, the antibodies were not verified in GPR39 knockout mice [165]. A role for ZnR/GPR39 in the regulation of the CREB/BDNF/TrkB (cyclic AMP response element binding protein/brain-derived neurotrophic factor/tyrosine receptor kinase B) pathway, and thereby in depression, has also been postulated, though it is not clear at present how Gαq signaling activates this pathway or whether these effects are lost in ZnR/GPR39 deficient mice [166,167].
5.5. Insulin Secretion
Pancreatic β-cells contain vesicular Zn2+ that is released together with insulin [168]. Several studies have highlighted a role for Zn2+ in the regulation of β-cell function and glucagon release [169,170,171]. The Zn2+ transporter ZnT8 is responsible for transporting Zn2+ into the insulin vesicles, and a mutation in this transporter of an Arg replacing Trp325 is associated with increased risk of developing type 2 diabetes [172]. Thus a role for ZnR/GPR39 in this tissue may have important physiological implications in the regulation of the Zn2+ releasing β-cells or neighboring cells within the islets of Langerhans. Knockout of ZnR/GPR39 does not immediately produce a phenotype under baseline conditions, and the knockout mice show normal insulin secretion. However, when fed a sucrose-rich diet, older mice show increased glucose levels and decreased insulin compared to the wild type [173]. Similarly, higher glucose levels were monitored in GPR39 knockout mice fed a high-fat diet [174]. In agreement, overexpression of ZnR/GPR39 in β-cells resulted in protection from streptozotocin-induced diabetes [175]. A recent study showed ZnR/GPR39 expression and Zn2+-dependent Ca2+ release in association with Zn2+-dependent insulin secretion [176]. Yet how ZnR/GPR39 activity regulates insulin secretion and whether this is an autocrine effect of endogenous Zn2+ released from the β-cells is still poorly understood.
5.6. Defects in Bone Composition
Zinc is accumulated in bone and plays a role in the dynamic maintenance of the structure of bones. Supplementation with dietary zinc enhances the strength of bones, but an underlying mechanism is not available. While several zinc transporters of the ZIP family have been associated with skeletal function [137], a role for ZnR/GPR39 was not described. Using GPR39 knockout mice, a recent study indicates that this receptor is important for osteoblast differentiation [61]. Hence, mice lacking ZnR/GPR39 showed impaired bone composition with decreased collagen content, likely involving ADAMTS metalloproteinase, which regulates collagen processing [61]. Most importantly, ZnR/GPR39 deficient osteoblasts showed lower ZIP13 expression, linking ZnR/GPR39 and Zn2+ transporters for the first time. Future studies aiming to determine how ZnR/GPR39 modulates Zn2+ transporters’ activity or expression can provide a more complete picture of the network of zinc homeostasis and its physiological implications.
5.7. ZnR/GPR39 in Cancer
Increased cell proliferation and migration, as well as the activation of MAPK and AKT, suggest a possible role for ZnR/GPR39 in cancer. Activation of ZnR/GPR39 signaling was monitored in androgen-independent, but not androgen-dependent, prostate cancer cells [59]. Extracellular Zn2+ via activation of ZnR/GPR39 in the prostate cancer cell line PC-3 enhances expression of S100A4 [84], a protein that is thought to enhance metastatic prostate cell proliferation and angiogenesis [177]. Other studies that associated ZnR/GPR39 expression in epithelial cells with cancer did not employ changes in extracellular or dietary Zn2+ to specifically study whether signaling pathway activation or Zn2+-dependent processes are affected in the cancer cells. These studies nevertheless indicate the importance of this receptor as a therapeutic target for cancer treatment. As such, GPR39 was overexpressed in primary human esophageal squamous cell carcinomas and its silencing reduced the tumorigenicity of these cells [178]. A recent study suggested that GPR39 expression is modulated in gastric adenocarcinoma [179], yet this study applied a previously incorrectly suggested ligand of GPR39 and not Zn2+ [180]. Interestingly, a link between the ZnR/GPR39 and mRNA levels of the Zn2+ transporter ZIP13 was recently shown in bone [61], but whether ZnR/GPR39 regulates other members of the ZIP family of Zn2+ transporters is unknown. Such a link between ZnR/GPR39 and ZIP transporters may further link the receptor to tumorigenesis. For example, ZIP6 and ZIP7 overexpression in breast cancer has been previously shown [181,182], and ZIP4 has recently been associated with ovarian stem cell growth and carcinoma [183]. Future studies aiming to specifically test the role of ZnR/GPR39 in cancer tissue and the link to ZIP transporters expression are of major interest and can provide a novel target for therapeutic tools.
6. Conclusions
ZnR/GPR39 is an important regulator of Zn2+-dependent signaling, functional in numerous epithelial cells, bone cells, and neurons—all tissues associated with Zn2+ homeostasis. Transient changes in extracellular Zn2+ occur during physiological activity and are sufficient to activate ZnR/GPR39. While dietary or serum zinc itself has been suggested to affect the physiological function or pathological conditions in these tissues, these changes in zinc concentration do not directly reflect local or cellular changes in the concentrations of the ionic Zn2+. In addition, Zn2+ interacts with a multitude of intracellular or extracellular proteins and modulates their activity, as described in the introduction; therefore, changes in Zn2+ concentration may affect many proteins and cellular functions and not just ZnR/GPR39 activity. Thus, this micronutrient is a poor therapeutic compound with inconsistent effects. However, elucidation of ZnR/GPR39 as a regulator of Zn2+-dependent cellular signaling can offer a novel handle to effective therapeutic approaches that will depend on ZnR/GPR39 agonists. Of note, ZnR/GPR39 is a member of the G-protein coupled receptor family, which is currently considered a major candidate for targeted therapies [184,185]. Finally, what regulates the activity of Zn2+ transporters is only partially understood; for example, it was previously shown that intracellular Zn2+ activation of metal-responsive elements regulates ZnT expression or that phosphorylation of ZIP regulates their expression [24,25]. In this context, a possible link between ZnR/GPR39 and the transporters may be a key to understanding Zn2+ homeostasis and is an important aim for future studies. Thus ZnR/GPR39 may serve as a specific and efficacious handle to modulate Zn2+ homeostatic proteins and signaling, thereby ameliorating physiological processes to enhance recovery.
Acknowledgments
Michal Hershfinkel was supported by Israeli Science Foundation grant #891/14 and Bi-National US Israel Science Foundation grant (2011126).
Conflicts of Interest
The author declares no conflict of interest.
References
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar] [PubMed]
- Prasad, A.S. Zinc in human health: Effect of zinc on immune cells. Mol. Med. 2008, 14, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H.; Frederickson, C.J.; Penland, J.G. History of zinc as related to brain function. J. Nutr. 2000, 130, 496S–502S. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, S.L.; McCormick, N.H.; Velasquez, V.; Lopez, V. Zinc in specialized secretory tissues: Roles in the pancreas, prostate, and mammary gland. Adv. Nutr. 2011, 2, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Weaver, B.P.; Andrews, G.K. The genetics of essential metal homeostasis during development. Genesis 2008, 46, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L. Zinc and carbonic anhydrase content of red cells in normals and in pernicious anemia. J. Clin. Investig. 1948, 27, 559. [Google Scholar] [PubMed]
- Vallee, B.L.; Altschule, M.D. Zinc in the mammalian organism, with particular reference to carbonic anhydrase. Physiol. Rev. 1949, 29, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc biochemistry, physiology, and homeostasis—Recent insights and current trends. BioMetals 2001, 14, 187–190. [Google Scholar] [CrossRef]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in cellular regulation: The nature and significance of “zinc signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Massarotti, A.; Hogstrand, C.; Maret, W. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics 2014, 6, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef] [PubMed]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Cousins, R.J. Mammalian zinc transporters. Annu. Rev. Nutr. 2004, 24, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Eide, D.J. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta 2006, 1763, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 2006, 11, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Mastroberardino, P.G. Metals and neurodegeneration. Neurobiol. Dis. 2015, 81, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Hasegawa, A.; Hojyo, S.; Ohashi, W.; Fukada, T.; Nishida, K.; Hirano, T. A novel role of the l-type calcium channel α1D subunit as a gatekeeper for intracellular zinc signaling: Zinc wave. PLoS ONE 2012, 7, e39654. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T.; Shimoda, S.; Ohashi, W.; Bin, B.H.; Koseki, H.; Hirano, T. The zinc transporter SLC39A14/ZIP14 controls G-protein coupled receptor-mediated signaling required for systemic growth. PLoS ONE 2011, 6, e18059. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Sitren, H.S.; Cousins, R.J. The zinc transporter Zip14 influences c-Met phosphorylation and hepatocyte proliferation during liver regeneration in mice. Gastroenterology 2012, 142, 1536–1546.e5. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-γ expression in activated human T cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Nimmanon, T.; Ziliotto, S.; Morris, S.; Flanagan, L.; Taylor, K.M. Phosphorylation of zinc channel ZIP7 drives MAPK, PI3K and mTOR growth and proliferation signalling. Metallomics 2017, 9, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci. Signal 2012, 5, ra11. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Hessels, A.M.; Taylor, K.M.; Merkx, M. Monitoring cytosolic and ER Zn2+ in stimulated breast cancer cells using genetically encoded FRET sensors. Metallomics 2016, 8, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takeda, T.A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Sullivan, P.G.; Sensi, S.L.; Steward, O.; Weiss, J.H. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 2001, 276, 47524–47529. [Google Scholar] [CrossRef] [PubMed]
- Reilly-O’Donnell, B.; Robertson, G.B.; Karumbi, A.; McIntyre, C.; Bal, W.; Nishi, M.; Takeshima, H.; Stewart, A.J.; Pitt, S.J. Dysregulated Zn2+ homeostasis impairs cardiac type-2 ryanodine receptor and mitsugumin 23 functions, leading to sarcoplasmic reticulum Ca2+ leakage. J. Biol. Chem. 2017, 292, 13361–13373. [Google Scholar] [CrossRef] [PubMed]
- Woodier, J.; Rainbow, R.D.; Stewart, A.J.; Pitt, S.J. Intracellular zinc modulates cardiac ryanodine receptor-mediated calcium release. J. Biol. Chem. 2015, 290, 17599–17610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maret, W. Zinc in the biosciences. Metallomics 2014, 6, 1174. [Google Scholar] [CrossRef] [PubMed]
- Kocyla, A.; Adamczyk, J.; Krezel, A. Interdependence of free zinc changes and protein complex assembly—Insights into zinc signal regulation. Metallomics 2017, 10, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M. Zinc, a dynamic signaling molecule. In Molecular Biology of Metal Homeostasis and Detoxification; Tamas, M., Martinoia, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 14, pp. 131–153. [Google Scholar]
- Frederickson, C.J.; Rampy, B.A.; Reamy Rampy, S.; Howell, G.A. Distribution of histochemically reactive zinc in the forebrain of the rat. J. Chem. Neuroanat. 1992, 5, 521–530. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Perez-Clausell, J.; Danscher, G. Zinc-containing 7S-NGF complex. Evidence from zinc histochemistry for localization in salivary secretory granules. J. Histochem. Cytochem. 1987, 35, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Danscher, G. Zinc-containing neurons in hippocampus and related CNS structures. Prog. Brain Res. 1990, 83, 71–84. [Google Scholar] [PubMed]
- Danscher, G.; Stoltenberg, M. Zinc-enriched neurons. J. Neurochem. 2003, 85, 10. [Google Scholar] [CrossRef]
- Ishii, K.; Sato, M.; Akita, M.; Tomita, H. Localization of zinc in the rat submandibular gland and the effect of its deficiency on salivary secretion. Ann. Otol. Rhinol. Laryngol. 1999, 108, 300–308. [Google Scholar] [CrossRef] [PubMed]
- McCormick, N.; Velasquez, V.; Finney, L.; Vogt, S.; Kelleher, S.L. X-ray fluorescence microscopy reveals accumulation and secretion of discrete intracellular zinc pools in the lactating mouse mammary gland. PLoS ONE 2010, 5, e11078. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Zinger, A.; Nevo, A.; Sekler, I.; Hershfinkel, M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J. Biol. Chem. 2010, 285, 26097–26106. [Google Scholar] [CrossRef] [PubMed]
- Hosie, A.M.; Dunne, E.L.; Harvey, R.J.; Smart, T.G. Zinc-mediated inhibition of GABAA receptors: Discrete binding sites underlie subtype specificity. Nat. Neurosci. 2003, 6, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wu, S.M. Modulation of glycine receptors in retinal ganglion cells by zinc. Proc. Natl. Acad. Sci. USA 1999, 96, 3234–3238. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.W.; Jacques, P.; Pierce, K.D.; Schofield, P.R. Zinc potentiation of the glycine receptor chloride channel is mediated by allosteric pathways. J. Neurochem. 1998, 71, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of nmda NR1-NR2A receptors. J. Neurosci. 1997, 17, 5711–5725. [Google Scholar] [PubMed]
- Herin, G.A.; Aizenman, E. Amino terminal domain regulation of NMDA receptor function. Eur. J. Pharmacol. 2004, 500, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Moran, A.; Hershfinkel, M.; Sekler, I. Inhibitory mechanism of store-operated Ca2+ channels by zinc. J. Biol. Chem. 2004, 279, 11106–11111. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.S.; King, B.F.; Burnstock, G. Modulatory activity of extracellular h+ and Zn2+ on ATP-responses at rP2X1 and rP2X3 receptors. Br. J. Pharmacol. 1999, 128, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castillo, C.; Morales, B.; Huidobro-Toro, J.P. Zinc and copper modulate differentially the P2X4 receptor. J. Neurochem. 2000, 74, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jung, Y.; Kim, D.; Koh, H.; Chung, J. Extracellular zinc activates p70 S6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J. Biol. Chem. 2000, 275, 25979–25984. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Park, K.S.; Kim, J.A.; Choi, K.Y. Differential modulation of zinc-stimulated p21(Cip/WAF1) and cyclin D1 induction by inhibition of PI3 kinase in HT-29 colorectal cancer cells. Exp. Mol. Med. 2002, 34, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M.; Moran, A.; Grossman, N.; Sekler, I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl. Acad. Sci. USA 2001, 98, 11749–11754. [Google Scholar] [CrossRef] [PubMed]
- Sunuwar, L.; Gilad, D.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, in health and disease. Front. Biosci. 2017, 22, 1469–1492. [Google Scholar]
- Hershfinkel, M. The zinc-sensing receptor, ZnR/GPR39: Signaling and significance. In Zinc Signals in Cellular Functions and Disorders; Fukada, T., Kambe, T., Eds.; Springer: Tokyo, Japan, 2014; pp. 11–134. [Google Scholar]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Hershfinkel, M. The extracellular zinc-sensing receptor mediates intercellular communication by inducing ATP release. Biochem. Biophys. Res. Commun. 2005, 332, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Dubi, N.; Gheber, L.; Fishman, D.; Sekler, I.; Hershfinkel, M. Extracellular zinc and zinc-citrate, acting through a putative zinc-sensing receptor, regulate growth and survival of prostate cancer cells. Carcinogenesis 2008, 29, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Egerod, K.L.; Jin, C.; Petersen, P.S.; Ostergaard, M.V.; Hald, J.; Sprinkel, A.M.; Storling, J.; Mandrup-Poulsen, T.; Holst, J.J.; et al. G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology 2009, 150, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Schmidt, F.; Guterman-Ram, G.; Khayyeri, H.; Jähn, K.; Hiram-Bab, S.; Orenbuch, A.; Katchkovsky, S.; Aflalo, A.; Isaksson, H.; et al. Perturbed bone composition and integrity with disorganized osteoblast function in zinc receptor/GPR39 deficient mice. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chorin, E.; Vinograd, O.; Fleidervish, I.; Gilad, D.; Herrmann, S.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 2011, 31, 12916–12926. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rosello, T.; Anderson, C.T.; Schopfer, F.J.; Zhao, Y.; Gilad, D.; Salvatore, S.R.; Freeman, B.A.; Hershfinkel, M.; Aizenman, E.; Tzounopoulos, T. Synaptic Zn2+ inhibits neurotransmitter release by promoting endocannabinoid synthesis. J. Neurosci. 2013, 33, 9259–9272. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Hildebrand, M.S.; Mullen, S.A.; Hildebrand, J.M.; Berkovic, S.F.; Petrou, S. Synaptic Zn2+ and febrile seizure susceptibility. Br. J. Pharmacol. 2017, 174, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iida, M.; Ando, M.; Nakamura, M.; Tamano, H.; Oku, N. Enhanced susceptibility to spontaneous seizures of noda epileptic rats by loss of synaptic Zn2+. PLoS ONE 2013, 8, e71372. [Google Scholar] [CrossRef] [PubMed]
- Nasehi, M.M.; Sakhaei, R.; Moosazadeh, M.; Aliramzany, M. Comparison of serum zinc levels among children with simple febrile seizure and control group: A systematic review. Iran. J. Child. Neurol. 2015, 9, 17–24. [Google Scholar] [PubMed]
- Ganesh, R.; Janakiraman, L. Serum zinc levels in children with simple febrile seizure. Clin. Pediatr. (Phila) 2008, 47, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Elsas, S.M.; Hazany, S.; Gregory, W.L.; Mody, I. Hippocampal zinc infusion delays the development of afterdischarges and seizures in a kindling model of epilepsy. Epilepsia 2009, 50, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Azriel-Tamir, H.; Arotsker, N.; Sekler, I.; Hershfinkel, M. Zinc sensing receptor signaling, mediated by GPR39, reduces butyrate-induced cell death in HT29 colonocytes via upregulation of clusterin. PLoS ONE 2012, 7, e35482. [Google Scholar]
- Moechars, D.; Depoortere, I.; Moreaux, B.; de Smet, B.; Goris, I.; Hoskens, L.; Daneels, G.; Kass, S.; Ver Donck, L.; Peeters, T.; et al. Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology 2006, 131, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Depoortere, I. Gi functions of GPR39: Novel biology. Curr. Opin. Pharmacol. 2012, 12, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Zhao, H.; Zhang, Y.; Peng, H.; Chen, Q.; Zhang, H.; Zheng, X.; Jin, Y.; Ni, H.; Duan, E.; et al. GPR39 is region-specifically expressed in mouse oviduct correlating with the Zn2+ distribution. Theriogenology 2017, 88, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tang, S.; Zhang, W.; Gao, W.; Chen, Y. GPR39 activates proliferation and differentiation of porcine intramuscular preadipocytes through targeting the PI3K/AKT cell signaling pathway. J. Recept. Signal Transduct. Res. 2016, 36, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, Y.; Zhang, X.; Chen, D. Zac1/GPR39 phosphorylating CaMK-II contributes to the distinct roles of Pax3 and Pax7 in myogenic progression. Biochim. Biophys. Acta 2018, 1864, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, G.L.; Alpers, D.H.; Binder, H.J.; Tran, C.D.; Ramakrishna, B.S.; Brown, I.; Manary, M.; Mortimer, E.; Young, G.P. The relevance of the colon to zinc nutrition. Nutrients 2015, 7, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Kirschke, C.P.; Huang, L. Immunohistochemical analysis of ZnT1, 4, 5, 6, and 7 in the mouse gastrointestinal tract. J. Histochem. Cytochem. 2007, 55, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M.; Silverman, W.F.; Sekler, I. The zinc sensing receptor, a link between zinc and cell signaling. Mol. Med. 2007, 13, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A novel Gαq/11-selective inhibitor. J. Biol. Chem. 2004, 279, 47438–47445. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Suzumura, K.; Nagai, K.; Kawasaki, T.; Takasaki, J.; Sekiguchi, M.; Moritani, Y.; Saito, T.; Hayashi, K.; Fujita, S.; et al. YM-254890 analogues, novel cyclic depsipeptides with Gαq/11 inhibitory activity from Chromobacterium sp. QS3666. Bioorg. Med. Chem. 2004, 12, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.H.; Brown, E.M. Disorders of calcium ion sensing. J. Clin. Endocrinol. Metab. 1996, 81, 2030–2035. [Google Scholar] [PubMed]
- Yasuda, S.; Miyazaki, T.; Munechika, K.; Yamashita, M.; Ikeda, Y.; Kamizono, A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J. Recept. Signal Transduct. Res. 2007, 27, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Stewart, A.J. GPR39: A Zn2+-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 2011, 68, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Asraf, H.; Salomon, S.; Nevo, A.; Sekler, I.; Mayer, D.; Hershfinkel, M. The ZnR/GPR39 interacts with the CaSR to enhance signaling in prostate and salivary epithelia. J. Cell. Physiol. 2013, 229, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.V.; Ren, P.G.; Avsian-Kretchmer, O.; Luo, C.W.; Rauch, R.; Klein, C.; Hsueh, A.J. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 2005, 310, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, B. Consideration in estimates of requirements and critical intake of zinc. Adaption, availability and interactions. Analyst 1995, 120, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.; Jensen, M.; Solgaard, P.; Sorensen, S.S.; Sandstrom, B. Zinc absorption estimated by fecal monitoring of zinc stable isotopes validated by comparison with whole-body retention of zinc radioisotopes in humans. J. Nutr. 1995, 125, 1274–1282. [Google Scholar] [PubMed]
- Sandstrom, B.; Cederblad, A.; Kivisto, B.; Stenquist, B.; Andersson, H. Retention of zinc and calcium from the human colon. Am. J. Clin. Nutr. 1986, 44, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Sekler, I.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, controls proliferation and differentiation of colonocytes and thereby tight junction formation in the colon. Cell Death Dis. 2014, 5, e1307. [Google Scholar] [CrossRef] [PubMed]
- Sunuwar, L.; Medini, M.; Cohen, L.; Sekler, I.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, triggers metabotropic calcium signalling in colonocytes and regulates occludin recovery in experimental colitis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- Sunuwar, L.; Asraf, H.; Donowitz, M.; Sekler, I.; Hershfinkel, M. The Zn2+-sensing receptor, ZnR/GPR39, upregulates colonocytic Cl− absorption, via basolateral KCC1, and reduces fluid loss. Biochim. Biophys. Acta 2017, 1863, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Albizu, L.; Balestre, M.N.; Breton, C.; Pin, J.P.; Manning, M.; Mouillac, B.; Barberis, C.; Durroux, T. Probing the existence of g protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol. Pharmacol. 2006, 70, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Frimurer, T.M.; Mende, F.; Graae, A.S.; Engelstoft, M.S.; Egerod, K.L.; Nygaard, R.; Gerlach, L.O.; Hansen, J.B.; Schwartz, T.W.; Holst, B. Model-based discovery of synthetic agonists for the Zn2+-sensing G-protein-coupled receptor 39 (GPR39) reveals novel biological functions. J. Med. Chem. 2017, 60, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/raf/MEK/ERK and PI3K/PTEN/AKT/MTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.P.; Rubinek, T.; Ryvo, L.; Wolf, I. Endocrine resistance in breast cancer: Focus on the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling pathway. Breast Care 2013, 8, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Weismann, D.; Ebert, S.; Adam, P.; Zink, M.; Beuschlein, F.; Hahner, S.; Allolio, B. AKT is highly phosphorylated in pheochromocytomas but not in benign adrenocortical tumors. J. Clin. Endocrinol. Metab. 2005, 90, 4366–4370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem. Biol. Interact. 2010, 185, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.C.; Waby, J.S.; Chirakkal, H.; Staton, C.A.; Corfe, B.M. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Mol. Cancer 2010, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Scharlau, D.; Borowicki, A.; Habermann, N.; Hofmann, T.; Klenow, S.; Miene, C.; Munjal, U.; Stein, K.; Glei, M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 2009, 682, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Butyrate and wnt signaling: A possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 2008, 7, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Opoka, W.; Adamek, D.; Plonka, M.; Reczynski, W.; Bas, B.; Drozdowicz, D.; Jagielski, P.; Sliwowski, Z.; Adamski, P.; Brzozowski, T. Importance of luminal and mucosal zinc in the mechanism of experimental gastric ulcer healing. J. Physiol. Pharmacol. 2010, 61, 581–591. [Google Scholar] [PubMed]
- Barceloux, D.G. Zinc. J. Toxicol. Clin. Toxicol. 1999, 37, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lansdown, A.B.; Mirastschijski, U.; Stubbs, N.; Scanlon, E.; Agren, M.S. Zinc in wound healing: Theoretical, experimental, and clinical aspects. Wound Repair Regen. 2007, 15, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Lansdown, A.B. Zinc in the healing wound. Lancet 1996, 347, 706–707. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Marsh, R.G.; Draelos, Z.D. Zinc and skin health: Overview of physiology and pharmacology. Dermatol. Surg. 2005, 31, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Dong, X.; Zhang, W. Obestatin changes proliferation, differentiation and apoptosis of porcine preadipocytes. Ann. Endocrinol. 2014, 75, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ganay, T.; Asraf, H.; Aizenman, E.; Bogdanovic, M.; Sekler, I.; Hershfinkel, M. Regulation of neuronal pH by the metabotropic zinc receptor mZnR/GPR39. J. Neurochem. 2015, 135, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.J.; Daly, C.A.; Huber, K.M. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J. Neurophysiol. 2006, 95, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Fibuch, E.E.; Mao, L. Regulation of mitogen-activated protein kinases by glutamate receptors. J. Neurochem. 2007, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.L.; Vasudevan, N.T.; Gupta, M.K.; Martelli, E.E.; Naga Prasad, S.V. G-protein coupled receptor resensitization-appreciating the balancing act of receptor function. Curr. Mol. Pharmacol. 2012, 5, 350–361. [Google Scholar] [CrossRef]
- Shimizu, Y.; Koyama, R.; Kawamoto, T. Rho kinase-dependent desensitization of GPR39; a unique mechanism of gpcr downregulation. Biochem. Pharmacol. 2017, 140, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Storjohann, L.; Holst, B.; Schwartz, T.W. Molecular mechanism of Zn2+ agonism in the extracellular domain of GPR39. FEBS Lett. 2008, 582, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Asraf, H.; Sekler, I.; Hershfinkel, M. Extracellular ph regulates zinc signaling via an ASP residue of the zinc-sensing receptor (ZnR/GPR39). J. Biol. Chem. 2012, 287, 33339–33350. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.S. The role of zinc in growth and cell proliferation. J. Nutr. 2000, 130, 1500S–1508S. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal luminal ph in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers. Compr. Physiol. 2011, 1, 2083–2100. [Google Scholar] [PubMed]
- Vaneckova, I.; Vylitova-Pletichova, M.; Beskid, S.; Zicha, J.; Pacha, J. Intracellular pH regulation in colonocytes of rat proximal colon. Biochim. Biophys. Acta 2001, 1536, 103–115. [Google Scholar] [CrossRef]
- Hachem, J.P.; Behne, M.; Aronchik, I.; Demerjian, M.; Feingold, K.R.; Elias, P.M.; Mauro, T.M. Extracellular ph controls NHE1 expression in epidermis and keratinocytes: Implications for barrier repair. J. Investig. Dermatol. 2005, 125, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Cardone, R.A.; Busco, G.; Krahling, H.; Schwab, A.; Reshkin, S.J. Protons extruded by NHE1: Digestive or glue? Eur. J. Cell. Biol. 2008, 87, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Panula, P.; Karhunen, T.; Heinonen, E. Fall in intracellular pH mediated by gabaa receptors in cultured rat astrocytes. Neurosci. Lett. 1991, 126, 9–12. [Google Scholar] [CrossRef]
- Manhas, N.; Shi, Y.; Taunton, J.; Sun, D. P90RSK activation contributes to cerebral ischemic damage via phosphorylation of Na+/H+ exchanger isoform 1. J. Neurochem. 2010, 114, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Aspects Med. 2013, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Jurd, R.; Moss, S.J. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 2010, 45, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Kaila, K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog. Brain Res. 2007, 160, 59–87. [Google Scholar] [PubMed]
- Viitanen, T.; Ruusuvuori, E.; Kaila, K.; Voipio, J. The K+-Cl− cotransporter KCC2 promotes gabaergic excitation in the mature rat hippocampus. J. Physiol. 2010, 588, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chen, C.X.; Liu, Y.J.; Aizenman, E.; Kandler, K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of gaba responses. Eur J. Neurosci. 2005, 21, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Karadsheh, M.; Delpire, E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 1999, 39, 558–568. [Google Scholar] [CrossRef]
- Saadi, R.A.; He, K.; Hartnett, K.A.; Kandler, K.; Hershfinkel, M.; Aizenman, E. Snare-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience 2012, 210, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Donowitz, M.; Verkman, A.S. Secretory diarrhoea: Mechanisms and emerging therapies. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Valenzano, M.C.; Mercado, J.M.; Zurbach, E.P.; Mullin, J.M. Zinc supplementation modifies tight junctions and alters barrier function of CACO-2 human intestinal epithelial layers. Dig. Dis. Sci. 2013, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Finamore, A.; Massimi, M.; Conti Devirgiliis, L.; Mengheri, E. Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J. Nutr. 2008, 138, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Lei, Z.; Wolf, P.G.; Gao, Y.; Guo, Y.M.; Zhang, B.K. Zinc supplementation, via GPR39, upregulates PKCζ to protect intestinal barrier integrity in Caco-2 cells challenged by salmonella enterica serovar typhimurium. J. Nutr. 2017, 147, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, Y.B.; Sekler, I.; Silverman, W.F. Histochemical and histofluorescence tracing of chelatable zinc in the developing mouse. J. Histochem. Cytochem. 2004, 52, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Takagishi, T.; Hara, T.; Fukada, T. Recent advances in the role of SLC39A/ZIP zinc transporters in vivo. Int. J. Mol. Sci. 2017, 18, 2708. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Lin, P.; Zhu, H.; Ko, J.K.; Hwang, M.; Tan, T.; Pan, Z.; Korichneva, I.; Ma, J. Zinc binding to MG53 protein facilitates repair of injury to cell membranes. J. Biol. Chem. 2015, 290, 13830–13839. [Google Scholar] [CrossRef] [PubMed]
- Krasovec, M.; Frenk, E. Acrodermatitis enteropathica secondary to crohn’s disease. Dermatology 1996, 193, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Sturniolo, G.C.; Fries, W.; Mazzon, E.; Di Leo, V.; Barollo, M.; D’Inca, R. Effect of zinc supplementation on intestinal permeability in experimental colitis. J. Lab. Clin. Med. 2002, 139, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.H.; Ko, J.K.; Fung, H.S.; Cho, C.H. Delineation of the protective action of zinc sulfate on ulcerative colitis in rats. Eur. J. Pharmacol. 2002, 443, 197–204. [Google Scholar] [CrossRef]
- Walker, C.L.; Black, R.E. Zinc for the treatment of diarrhoea: Effect on diarrhoea morbidity, mortality and incidence of future episodes. Int. J. Epidemiol. 2010, 39, i63–i69. [Google Scholar] [CrossRef] [PubMed]
- Alam, D.S.; Yunus, M.; El Arifeen, S.; Chowdury, H.R.; Larson, C.P.; Sack, D.A.; Baqui, A.H.; Black, R.E. Zinc treatment for 5 or 10 days is equally efficacious in preventing diarrhea in the subsequent 3 months among bangladeshi children. J. Nutr. 2011, 141, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Yang, J.; Chen, T.E.; Zachos, N.C.; Kovbasnjuk, O.; Verkman, A.S.; Donowitz, M. Translating molecular physiology of intestinal transport into pharmacologic treatment of diarrhea: Stimulation of Na+ absorption. Clin. Gastroenterol. Hepatol. 2014, 12, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Di Sole, F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am. J. Physiol. Cell Physiol. 2012, 302, C1569–C1587. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Ko, E.A.; Tradtrantip, L.; Donowitz, M.; Verkman, A.S. Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin. Gastroenterol. Hepatol. 2014, 12, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Passariello, A.; Terrin, G.; De Marco, G.; Cecere, G.; Ruotolo, S.; Marino, A.; Cosenza, L.; Tardi, M.; Nocerino, R.; Berni Canani, R. Efficacy of a new hypotonic oral rehydration solution containing zinc and prebiotics in the treatment of childhood acute diarrhea: A randomized controlled trial. J. Pediatr. 2011, 158, 288–292.e281. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Phillips, A.M.; Mullen, S.A.; Adlard, P.A.; Hardies, K.; Damiano, J.A.; Wimmer, V.; Bellows, S.T.; McMahon, J.M.; Burgess, R.; et al. Loss of synaptic Zn2+ transporter function increases risk of febrile seizures. Sci. Rep. 2015, 5, 17816. [Google Scholar] [CrossRef] [PubMed]
- Farahani, H.N.; Ashthiani, A.R.; Masihi, M.S. Study on serum zinc and selenium levels in epileptic patients. Neurosciences 2013, 18, 138–142. [Google Scholar] [PubMed]
- Mitsuya, K.; Nitta, N.; Suzuki, F. Persistent zinc depletion in the mossy fiber terminals in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Epilepsia 2009, 50, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Xu, K.; Yoo, J.; Chen, T.T.; Andrews, G.; Noebels, J.L. Knockout of Zn transporters ZIP-1 and ZIP-3 attenuates seizure-induced ca1 neurodegeneration. J. Neurosci. 2011, 31, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Robbins, C.A.; Wenzel, H.J.; Schwartzkroin, P.A.; Palmiter, R.D. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000, 39, 153–169. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lovinger, D.; Delpire, E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol. 2005, 93, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Woo, N.S.; Lu, J.; England, R.; McClellan, R.; Dufour, S.; Mount, D.B.; Deutch, A.Y.; Lovinger, D.M.; Delpire, E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 2002, 12, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Gilad, D.; Shorer, S.; Ketzef, M.; Friedman, A.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol. Dis. 2015, 81, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Q.; Yao, H.; Haddad, G.G. Increased neuronal excitability and seizures in the Na+/H+ exchanger null mutant mouse. Am. J. Physiol. Cell Physiol. 2001, 281, C496–C503. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhao, P.; Xue, J.; Gu, X.Q.; Sun, X.; Yao, H.; Haddad, G.G. Na+ channel expression and neuronal function in the Na+/H+ exchanger 1 null mutant mouse. J. Neurophysiol. 2003, 89, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced alzheimer’s Aβ1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Tempaku, M.; Sasaki, M.; Uematsu, C.; Sato, S.; Kanazawa, H.; Datki, Z.L.; Adlard, P.A.; Bush, A.I. Extracellular Zn2+ is essential for amyloid β1-42-induced cognitive decline in the normal brain and its rescue. J. Neurosci. 2017, 37, 7253–7262. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch-Dahan, C.; Asraf, H.; Bogdanovic, M.; Sekler, I.; Bush, A.I.; Hershfinkel, M. Amyloid β attenuates metabotropic zinc sensing receptor, mZnR/GPR39, dependent Ca2+, ERK1/2 and clusterin signaling in neurons. J. Neurochem. 2016, 139, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, M.A.; Kranz, T.M.; Kleinhaus, K.; Joe, P.; Getz, M.; Johnson, P.; Chao, M.V.; Malaspina, D. The emerging role for zinc in depression and psychosis. Front. Pharmacol. 2017, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Doboszewska, U.; Szewczyk, B.; Sowa-Kucma, M.; Misztak, P.; Piekoszewski, W.; Trela, F.; Ostachowicz, B.; Nowak, G. The involvement of the GPR39-Zn2+-sensing receptor in the pathophysiology of depression. Studies in rodent models and suicide victims. Neuropharmacology 2014, 79, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Nowak, G. GPR39 up-regulation after selective antidepressants. Neurochem. Int. 2013, 62, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Gawel, M.; Librowski, T.; Reczynski, W.; Bystrowska, B.; Holst, B. Investigation of the GPR39 zinc receptor following inhibition of monoaminergic neurotransmission and potentialization of glutamatergic neurotransmission. Brain Res. Bull. 2015, 115, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cichy, A.; Sowa-Kucma, M.; Legutko, B.; Pomierny-Chamiolo, L.; Siwek, A.; Piotrowska, A.; Szewczyk, B.; Poleszak, E.; Pilc, A.; Nowak, G. Zinc-induced adaptive changes in NMDA/glutamatergic and serotonergic receptors. Pharmacol. Rep. 2009, 61, 1184–1191. [Google Scholar] [CrossRef]
- Mlyniec, K.; Nowak, G. Up-regulation of the GPR39 Zn2+-sensing receptor and CREB/BDNF/TrkB pathway after chronic but not acute antidepressant treatment in the frontal cortex of zinc-deficient mice. Pharmacol. Rep. 2015, 67, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Gee, K.R.; Kennedy, R.T. Imaging of Zn2+ release from pancreatic β-cells at the level of single exocytotic events. Anal. Chem. 2003, 75, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Bloc, A.; Cens, T.; Cruz, H.; Dunant, Y. Zinc-induced changes in ionic currents of clonal rat pancreatic β-cells: Activation of ATP-sensitive K+ channels. J. Physiol. (Lond.) 2000, 529, 723–734. [Google Scholar] [CrossRef]
- Ferrer, R.; Soria, B.; Dawson, C.M.; Atwater, I.; Rojas, E. Effects of Zn2+ on glucose-induced electrical activity and insulin release from mouse pancreatic islets. Am. J. Physiol. 1984, 246, C520–C527. [Google Scholar] [CrossRef] [PubMed]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. Beta-cell secretory products activate α-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in pancreatic islet biology, insulin sensitivity, and diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Richard, A.M.; Will, S.; Syed, J.; Stedman, N.; Perreault, M.; Gimeno, R.E. Disruption of G protein-coupled receptor 39 impairs insulin secretion in vivo. Endocrinology 2009, 150, 2586–2595. [Google Scholar] [CrossRef] [PubMed]
- Verhulst, P.J.; Lintermans, A.; Janssen, S.; Loeckx, D.; Himmelreich, U.; Buyse, J.; Tack, J.; Depoortere, I. GPR39, a receptor of the ghrelin receptor family, plays a role in the regulation of glucose homeostasis in a mouse model of early onset diet-induced obesity. J. Neuroendocrinol. 2011, 23, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Egerod, K.L.; Jin, C.; Petersen, P.S.; Wierup, N.; Sundler, F.; Holst, B.; Schwartz, T.W. Beta-cell specific overexpression of GPR39 protects against streptozotocin-induced hyperglycemia. Int. J. Endocrinol. 2011, 2011, 401258. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.M.; Abdel-Wahab, Y.H.; Vasu, S.; Flatt, P.R.; McKillop, A.M. GPR39 receptors and actions of trace metals on pancreatic β cell function and glucose homoeostasis. Acta Diabetol. 2016, 53, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, H.; Zhu, Y.H.; Qin, Y.R.; Dai, Y.; Zeng, T.; Chen, L.; Nie, C.; Tang, H.; Li, Y.; et al. Overexpression of GPR39 contributes to malignant development of human esophageal squamous cell carcinoma. BMC Cancer 2011, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alen, B.O.; Leal-Lopez, S.; Alen, M.O.; Viano, P.; Garcia-Castro, V.; Mosteiro, C.S.; Beiras, A.; Casanueva, F.F.; Gallego, R.; Garcia-Caballero, T.; et al. The role of the obestatin/GPR39 system in human gastric adenocarcinomas. Oncotarget 2016, 7, 5957–5971. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Egerod, K.L.; Schild, E.; Vickers, S.P.; Cheetham, S.; Gerlach, L.O.; Storjohann, L.; Stidsen, C.E.; Jones, R.; Beck-Sickinger, A.G.; et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 2007, 148, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M. Liv-1 breast cancer protein belongs to new family of histidine-rich membrane proteins with potential to control intracellular Zn2+ homeostasis. IUBMB Life 2000, 49, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M. A distinct role in breast cancer for two liv-1 family zinc transporters. Biochem. Soc. Trans. 2008, 36, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Li, P.; Wang, W.; Wang, J.; Gerry, E.; Wang, T.L.; Shih, I.M.; Nephew, K.P.; Xu, Y. The novel ZIP4 regulation and its role in ovarian cancer. Oncotarget 2017, 8, 90090–90107. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Custodi, C.; Nuti, R.; Oprea, T.I.; Macchiarulo, A. Fitting the complexity of GPCRs modulation into simple hypotheses of ligand design. J. Mol. Graph. Model. 2012, 38, 70–81. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representations of common Zn2+ sensing receptor, ZnR/GPR39, signaling in epithelial cells. Extracellular signal–regulated kinases, ERK; Phosphatidylinositol-4,5-bisphosphate 3 (PI3) kinase/AKT, PI3K/AKT; Phospholipase C, PLC.
Figure 1.
Schematic representations of common Zn2+ sensing receptor, ZnR/GPR39, signaling in epithelial cells. Extracellular signal–regulated kinases, ERK; Phosphatidylinositol-4,5-bisphosphate 3 (PI3) kinase/AKT, PI3K/AKT; Phospholipase C, PLC.

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hershfinkel, M. The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. Int. J. Mol. Sci. 2018, 19, 439. https://doi.org/10.3390/ijms19020439
AMA Style
Hershfinkel M. The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. International Journal of Molecular Sciences. 2018; 19(2):439. https://doi.org/10.3390/ijms19020439
Chicago/Turabian StyleHershfinkel, Michal. 2018. "The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease" International Journal of Molecular Sciences 19, no. 2: 439. https://doi.org/10.3390/ijms19020439
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.