The T2238C Human Atrial Natriuretic Peptide Molecular Variant and the Risk of Cardiovascular Diseases


Abstract

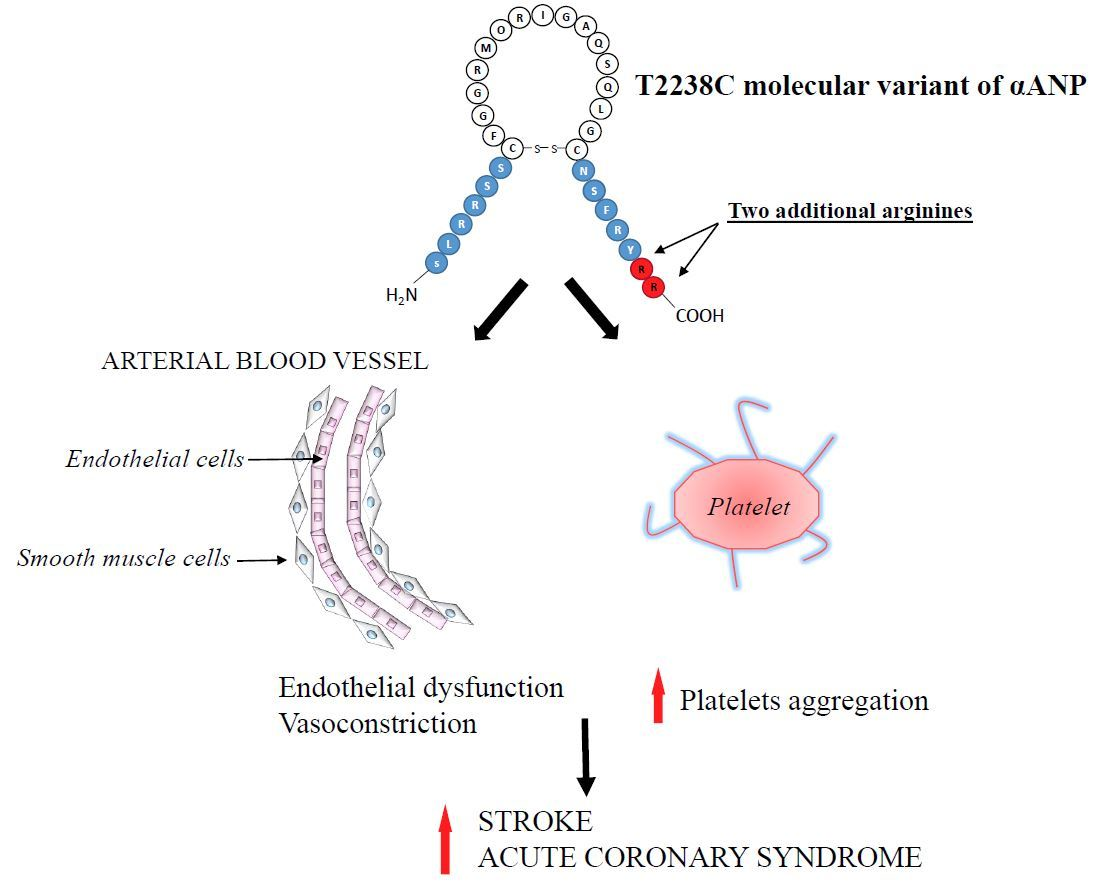{kind=link}
{kind=link}
1. Introduction
1.1. Clinical Evidence of the Pathological Role of the T2238C/ANP Molecular Variant
1.2. Discovery of the Molecular Mechanisms Underlying the Deleterious Effects of CC2238/αΑnp
2. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of interest
References
- Levin, E.R.; Gardner, D.G.; Samson, W.K. Natriuretic peptides. N. Engl. J. Med. 1998, 339, 321–328. [Google Scholar] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol. 2009, 191, 341–366. [Google Scholar]
- Rubattu, S.; Sciarretta, S.; Valenti, V.; Stanzione, R.; Volpe, M. Natriuretic peptides: An update on bioactivity, potential therapeutic use and implication in cardiovascular diseases. Am. J. Hypertens. 2008, 21, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.G.; Deschepper, C.F.; Ganong, W.F.; Hane, S.; Fiddes, J.; Baxter, J.D.; Lewicki, J. Extra-atrial expression of the gene for atrial natriuretic factor. Proc. Natl. Acad. Sci. USA 1986, 83, 6697–6701. [Google Scholar] [CrossRef] [PubMed]
- Nemer, M.; Chamberland, M.; Sirois, D.; Argentin, S.; Drouin, J.; Dixon, R.A.; Zivin, R.A.; Condra, J.H. Gene structure of human cardiac hormone precursor, pronatriodilatin. Nature 1984, 312, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cao, P.; Dong, N.; Peng, J.; Zhang, C.; Wang, H.; Zhou, T.; Yang, J.; Zhang, Y.; Martelli, E.E.; et al. PCSK6-mediated corin activation is essential for normal blood pressure. Nat. Med. 2015, 21, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Rubattu, S. Novel insights into the mechanisms regulating pro-atrial natriuretic peptide cleavage in the heart and blood pressure regulation. Proprotein Convertase Subtilisin/Kexin 6 is the corin activating enzyme. Circ. Res. 2016, 118, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Vesely, D.L. Atrial natriuretic hormones originating from the N-terminus of the atrial natriureticfactor prohormone. Clin. Exp. Pharmacol. Physiol. 1995, 22, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Sciarretta, S.; Volpe, M. Atrial natriuretic peptide gene variants and circulating levels: Implications in cardiovascular diseases. Clin. Sci. 2014, 127, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Volpe, M.; Kreutz, R.; Ganten, U.; Ganten, D.; Lindpaintner, K. Chromosomal mapping of genetic loci contributing to stroke in an animal model of a complex human disease. Nat. Genet. 1996, 13, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Lee, M.A.; De Paolis, P.; Giliberti, R.; Gigante, B.; Lombardi, A.; Volpe, M.; Lindpaintner, K. Altered structure, regulation and function of the gene encoding atrial natriuretic peptide in the stroke-prone spontaneously hypertensive rat. Circ. Res. 1999, 85, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Giliberti, R.; De Paolis, P.; Stanzione, R.; Spinsanti, P.; Volpe, M. Effect of a regulatory mutation on the rat atrial natriuretic peptide gene transcription. Peptides 2002, 23, 555–560. [Google Scholar] [CrossRef]
- De Paolis, P.; Nobili, V.; Lombardi, A.; Tarasi, D.; Barbato, D.; Ganten, U.; Brunetti, E.; Volpe, M.; Rubattu, S. Role of a molecular variant of the rat atrial natriuretic peptide gene on vascular remodeling. Ann. Clin. Lab. Sci. 2007, 37, 135–140. [Google Scholar] [PubMed]
- Rubattu, S.; Giliberti, R.; Ganten, U.; Volpe, M. A differential brain ANP expression cosegregates with occurrence of early stroke in the stroke-prone phenotype of the spontaneously hypertensive rat. J. Hypertens. 1999, 17, 1849–1852. [Google Scholar] [CrossRef] [PubMed]
- Gruchala, M.; Ciecwierz, D.; Wasag, B.; Targonski, R.; Dubaniewicz, W.; Nowak, A.; Sobiczewski, W.; Ochman, K.; Romanowski, P.; Limon, J.; et al. Association of the ScaI atrial natriuretic peptide gene polymorphism with nonfatal myocardial infarction and extent of coronary artery disease. Am. Heart J. 2003, 145, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Stanzione, R.; Di Angelantonio, E.; Zanda, B.; Evangelista, A.; Tarasi, D.; Gigante, B.; Pirisi, A.; Brunetti, E.; Volpe, M. Atrial natriuretic peptide gene polymorphisms and the risk of ischemic stroke in humans. Stroke 2004, 35, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, L.; He, M.; Hu, B.; Wu, T. ANP T2238C, C-664G gene polymorphism and coronary heart disease in Chinese population. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Barbato, E.; Bartunek, J.; Mangiacapra, F.; Sciarretta, S.; Stanzione, R.; Delrue, L.; Cotugno, M.; Marchitti, S.; Iaccarino, G.; Sirico, G.; et al. Influence of rs5065 atrial natriuretic peptide gene variant on coronary artery disease. J. Am. Coll. Cardiol. 2012, 59, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; De Giusti, M.; Farcomeni, A.; Abbolito, S.; Comito, F.; Cangianiello, S.; Squillace Greco, E.; Dito, E.; Pagliaro, B.; Cotugno, M.; et al. T2238C ANP gene variant and risk of recurrent acute coronary syndromes in an Italian cohort of ischemic heart disease patients. J. Cardiovasc. Med. 2016, 17, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Ziaee, S.; Kalayinia, S.; Boroumand, M.A.; Pourgholi, L.; Cheraghi, S.; Anvari, M.S.; Sheikhvatan, M. Association between the atrial natriuretic peptide rs5065 gene polymorphism and the presence and severity of coronary artery disease in an Iranian population. Coron. Artery Dis. 2014, 25, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.H.; Go, H.Y.; Park, S.; Shin, Y.C.; Kim, S.H.; Ko, S.G. Potential association between frequent nonsynonymous variant of NPPA and cardioembolic stroke. DNA Cell Biol. 2012, 31, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Larifla, L.; Rambhojan, C.; Joannes, M.O.; Maimaitiming-Madani, S.; Donnet, J.P.; Marianne-Pepin, T.; Chout, R.; Roussel, R.; Foucan, L. Gene polymorphisms of FABP2, ADIPOQ and ANP and the risk of hypertriglyceridemia and methabolic syndrome in Afro-Caribbeans. PLoS ONE 2016, 11, e0163421. [Google Scholar] [CrossRef] [PubMed]
- Roussel, R.; Tregouet, D.A.; Hadjadj, S.; Jeunemaitre, X.; Marre, M. Investigation of the human ANP gene in type 1 diabetic nephropathy: Case-control and follow-up studies. Diabetes 2004, 53, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Cannone, V.; Huntley, B.K.; Olson, T.M.; Heublein, D.M.; Scott, C.G.; Bailey, K.R.; Redfield, M.M.; Rodeheffer, R.J.; Burnett, J.C., Jr. Atrial natriuretic genetic variant rs5065 and risk for cardiovascular disease in the general community: A 9-year follow-up study. Hypertension 2013, 62, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Francia, P.; Ricotta, A.; Frattari, A.; Stanzione, R.; Modestino, A.; Mercanti, F.; Adduci, C.; Sensini, I.; Cotugno, M.; Balla, C.; et al. Atrial natriuretic peptide single nucleotide polymorphisms in patients with non-familial structural atrial fibrillation. Clin. Med. Insights Cardiol. 2013, 7, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Siebert, J.; Lewicki, Ł.; Myśliwska, J.; Młotkowska, M.; Rogowski, J. ScaI atrial natriuretic peptide gene polymorphisms and their possible association with postoperative atrial fibrillation—A preliminary report. Arch. Med. Sci. 2017, 13, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Niu, W. The relationship between natriuretic peptide precursor a gene T2238C polymorphism and hypertension: A meta-analysis. Int. J. Hypertens. 2011, 653698. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.I.; Boerwinkle, E.; Davis, B.R.; Ford, C.E.; Eckfeldt, J.H.; Leiendecker-Foster, C.; Arnett, D.K. Pharmacogenetic association of the NPPA T2238C genetic variant with cardiovascular disease outcomes in patients with hypertension. JAMA 2008, 299, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Tse, M.Y.; Pang, S.C.; McLellan, C.S.; King, W.D.; Johri, A.M. Sex differences of the natriuretic peptide polymorphism associated with angiographic coronaty atherosclerosis. Cardiol. Res. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Larifla, L.; Maimaitiming, S.; Velayoudom-Cephise, F.L.; Ferdinand, S.; Blanchet-Deverly, A.; Benabdallah, S.; Donnet, J.P.; Atallah, A.; Roussel, R.; Foucan, L. Association of 2238T>C polymorphism of the atrial natriuretic peptide gene with coronary artery disease in Afro-Caribbeans with type 2 diabetes. Am. J. Hypertens. 2012, 25, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Marchitti, S.; Stanzione, R.; Evangelista, A.; Di Castro, S.; Savoia, C.; Quarta, G.; Sciarretta, S.; Ruco, L.; Volpe, M.; et al. ROS-mediated differential effects of the human atrial natriuretic peptide T2238C genetic variant on endothelial cells in vitro. J. Hypertens. 2009, 27, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Marchitti, S.; Bianchi, F.; Moyes, A.; Barbato, E.; Di Castro, S.; Stanzione, R.; Cotugno, M.; Castello, L.; Calvieri, C.; et al. The C2238 atrial natriuretic peptide molecular variant is associated with endothelial damage and dysfunction through natriuretic peptide receptor C signaling. Circ. Res. 2013, 112, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Sciarretta, S.; Morriello, A.; Calvieri, C.; Battistoni, A.; Volpe, M. NPR-C: A component of the natriuretic peptide family with implications in human diseases. J. Mol. Med. 2010, 88, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Bellis, A.; Castaldo, D.; Trimarco, V.; Monti, M.G.; Chivasso, P.; Sadoshima, J.; Trimarco, B.; Morisco, C. Crosstalk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Marchitti, S.; Bianchi, F.; Di Castro, S.; Stanzione, R.; Cotugno, M.; Bozzao, C.; Sciarretta, S.; Volpe, M. The C2238/αANP variant is a negative modulator of both viability and function of coronary artery smooth muscle cells. PLoS ONE 2014, 9, e113108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Trans. Res. 2010, 3, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointima formation. Circ. Res. 2007, 100, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Garofalo, M.; Croce, C.M.; Condorelli, G. ApoptomiRs in vascular cells: Their role in physiological and pathological angiogenesis. Vasc. Pharmacol. 2011, 55, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Song, J.T.; Hu, B.; Yan Qu, H.; Bi, C.I.; Huang, X.Z. Mechanical stretch modulates MicroRNA 21 expression, participating in proliferation and apoptosis in cultured human aortic smooth muscle cells. PLoS ONE 2012, 7, e47657. [Google Scholar] [CrossRef] [PubMed]
- Kotlo, K.U.; Hesabi, B.; Danziger, R.S. Implication of microRNAs in atrial natriuretic peptide and nitric oxide signaling in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2011, 301, C929–C937. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Hussain, S.R.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatide and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Sun, H.; Kerfant, B.G.; Crackower, M.A.; Penninger, J.M.; Backx, P.H. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 2004, 37, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Y.; Shan, H.; Pan, Z.; Li, X.; Li, B.; Xu, C.; Zhang, B.; Zhang, F.; Dong, D.; et al. MicroRNA-1 downregulation by propanolol in a rat model of myocardial infarction: A new mechanism for ischaemic cardioprotection. Cardiovasc. Res. 2009, 84, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Zhou, Y.; Zhu, W.; Zhang, H.; Wu, S.; Chen, J.; Yan, G. MicroRNA 335 is required for differentiation of malignant glioma cells induced by activation of cAMP/Protein Kinase A pathway. Mol. Pharmacol. 2012, 81, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Stanzione, R.; Sciarretta, S.; Marchitti, S.; Bianchi, F.; Di Castro, S.; Scarpino, S.; Cotugno, M.; Frati, G.; Volpe, M.; Rubattu, S. C2238/αANP modulates Apolipoprotein E through Egr-1/miR199a in vascular smooth muscle cells in vitro. Cell Death Dis. 2015, 6, e2033. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vita, J.; Sanchez-Galan, E.; Santamaria, B.; Sanchez-Lopez, E.; Rodrigues-Diez, R.; Blanco-Colio, L.M.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M. Essential role of TGF-β/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS ONE 2008, 3, e3959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dai, P.; Cheng, D.; Zhang, L.; Chen, Z.; Meng, X.; Zhang, F.; Han, X.; Liu, J.; Pan, J.; et al. Obesity occurring in apolipoprotein E-knockout mice has mild effects on fertility. Reproduction 2013, 147, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.H.; Duthie, S.J.; Kwun, I.; Ha, T.; Gordon, M.J. Rapid quantification of aortic lesions in ApoE−/− mice. J. Vasc. Res. 2009, 46, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.; Mitrou, P.N.; Bowman, R.; Luben, R.; Wareham, N.J.; Khaw, K.T.; Bingham, S. APOE genotype, lipids, and coronary heart disease risk: A prospective population study. Arch. Intern. Med. 2009, 169, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P. Platelet oxidative stress and thrombosis. Thromb. Res. 2012, 129, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Loeb, A.L.; Gear, A.R. Potentiation of platelet aggregation by atrial natriuretic peptide. Life Sci. 1988, 43, 731–738. [Google Scholar] [CrossRef]
- Carnevale, R.; Pignatelli, P.; Frati, G.; Nocella, C.; Stanzione, R.; Pastori, D.; Marchitti, S.; Valenti, V.; Santulli, M.; Barbato, E.; et al. C2238 ANP gene variant promotes increased platelet aggregation through the activation of Nox2 and the reduction of cAMP. Sci. Rep. 2017, 7, 3797. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. Gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, T.; Barbato, E.; De Biase, C.; Di Gioia, G.; Cotugno, M.; Stanzione, R.; Trimarco, B.; Sciarretta, S.; Volpe, M.; Wijns, W.; et al. T2238C atrial natriuretic peptide gene variant and the response to antiplatelet therapy in stable ischemic heart disease patients. J. Cardiovasc. Transl. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care 2009, 32, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Sciarretta, S.; Marchitti, S.; D’Agostino, M.; Battistoni, A.; Calvieri, C.; Volpe, M. NT-proANP/ANP is a determinant of vascular damage in humans. High Blood Press Cardiovasc. Prev. 2010, 17, 121–130. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubattu, S.; Sciarretta, S.; Marchitti, S.; Bianchi, F.; Forte, M.; Volpe, M. The T2238C Human Atrial Natriuretic Peptide Molecular Variant and the Risk of Cardiovascular Diseases. Int. J. Mol. Sci. 2018, 19, 540. https://doi.org/10.3390/ijms19020540
Rubattu S, Sciarretta S, Marchitti S, Bianchi F, Forte M, Volpe M. The T2238C Human Atrial Natriuretic Peptide Molecular Variant and the Risk of Cardiovascular Diseases. International Journal of Molecular Sciences. 2018; 19(2):540. https://doi.org/10.3390/ijms19020540
Chicago/Turabian StyleRubattu, Speranza, Sebastiano Sciarretta, Simona Marchitti, Franca Bianchi, Maurizio Forte, and Massimo Volpe. 2018. "The T2238C Human Atrial Natriuretic Peptide Molecular Variant and the Risk of Cardiovascular Diseases" International Journal of Molecular Sciences 19, no. 2: 540. https://doi.org/10.3390/ijms19020540
APA StyleRubattu, S., Sciarretta, S., Marchitti, S., Bianchi, F., Forte, M., & Volpe, M. (2018). The T2238C Human Atrial Natriuretic Peptide Molecular Variant and the Risk of Cardiovascular Diseases. International Journal of Molecular Sciences, 19(2), 540. https://doi.org/10.3390/ijms19020540