Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles

 , ,
, ,
Abstract
:1. Introduction
2. Results
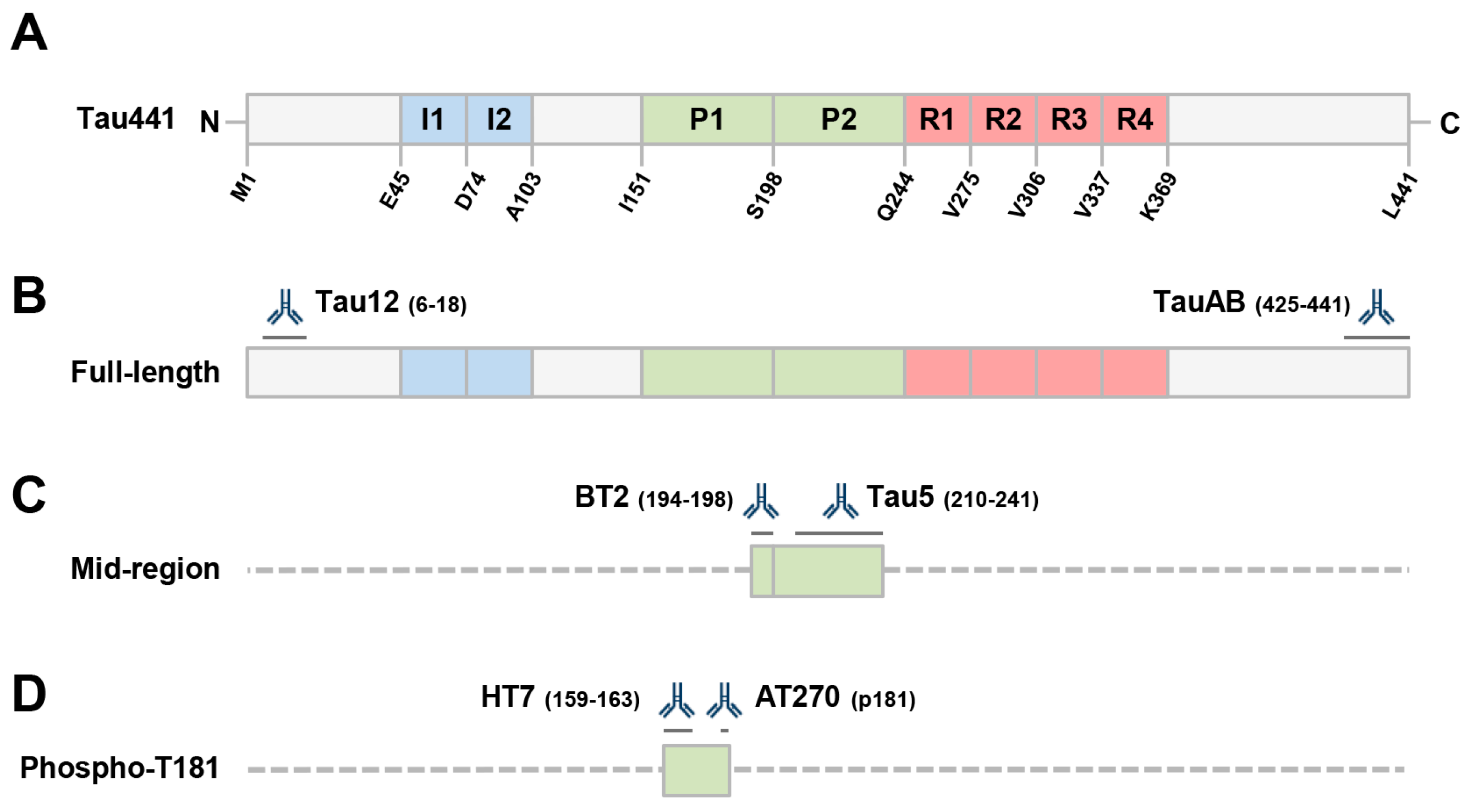
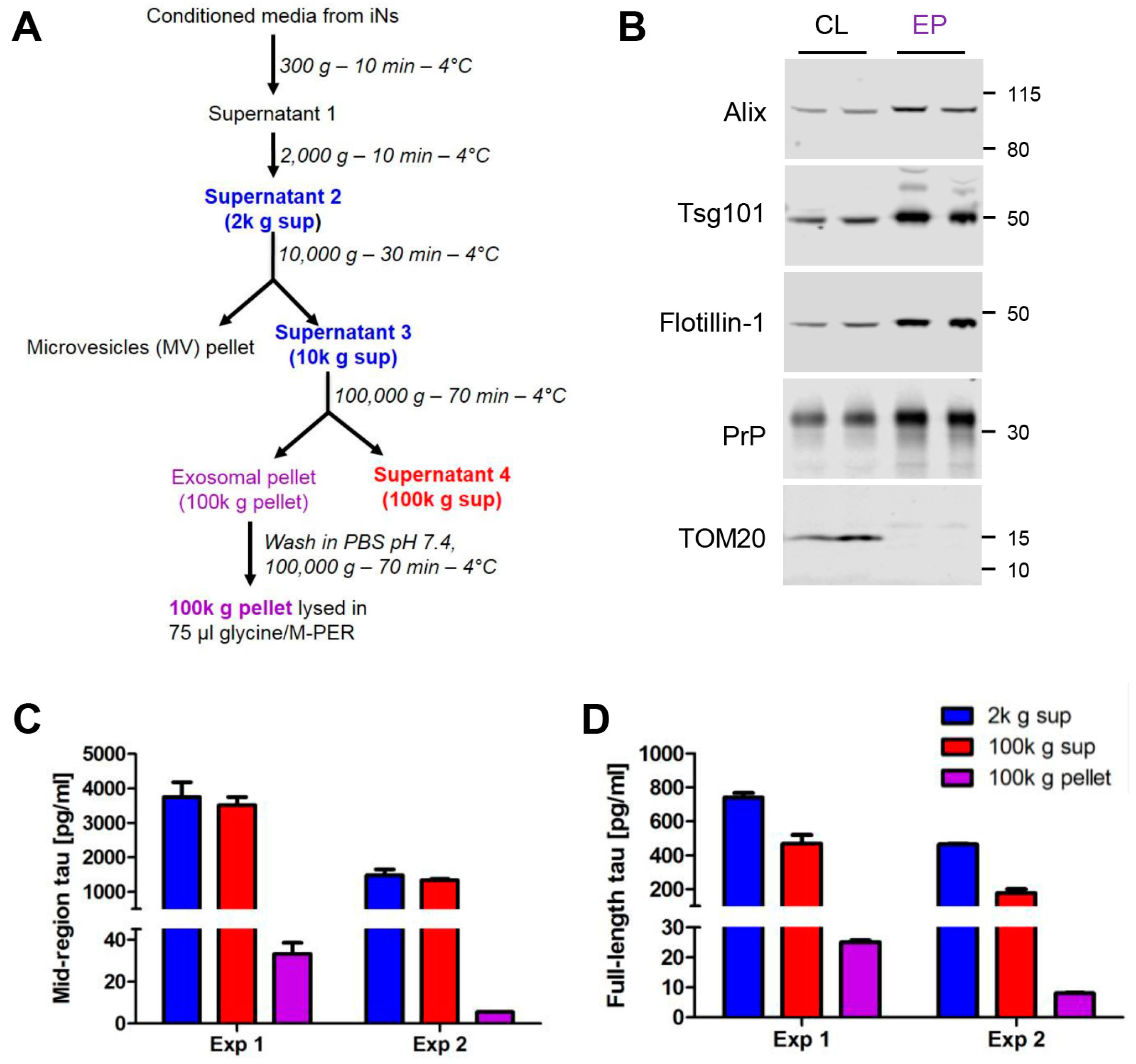
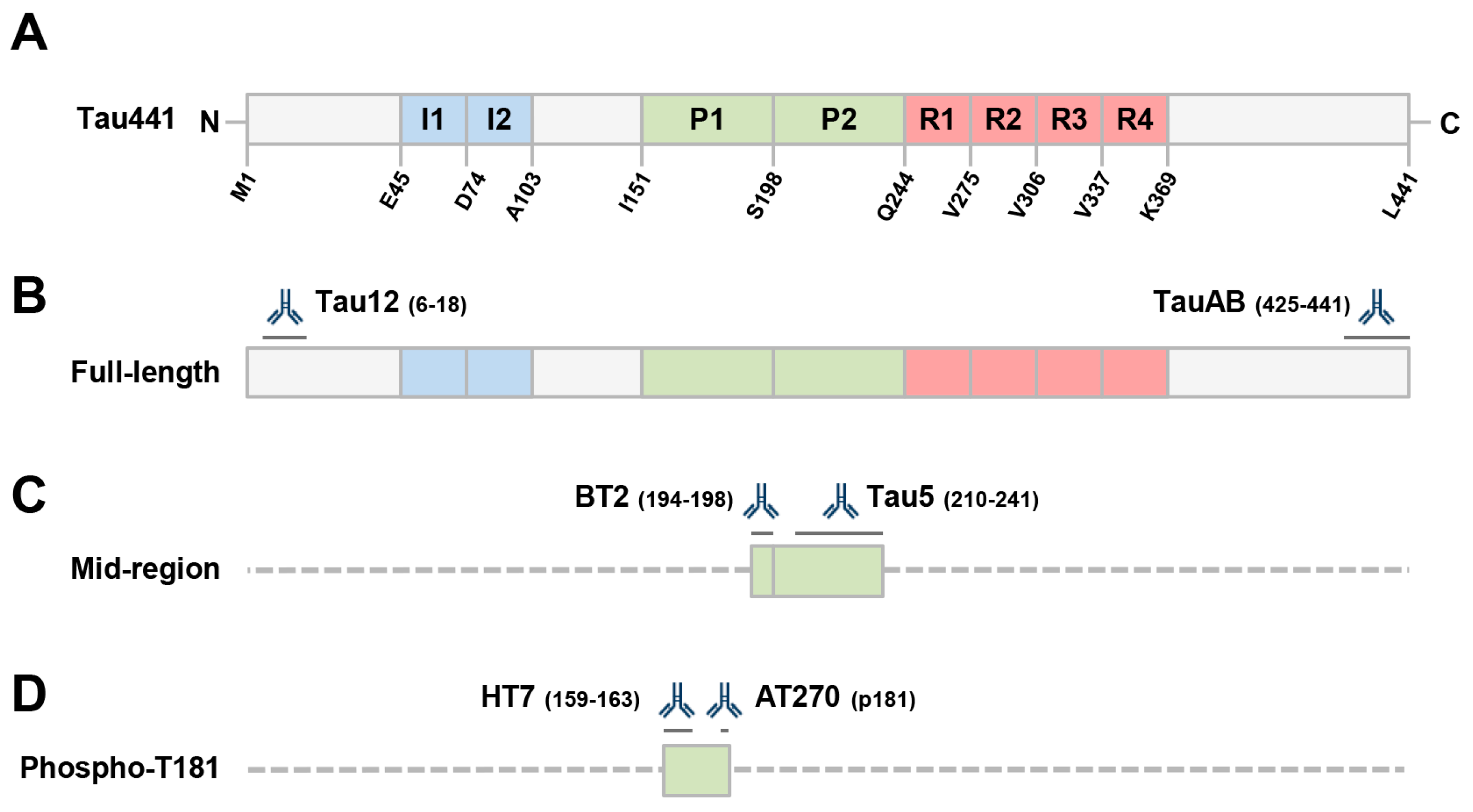
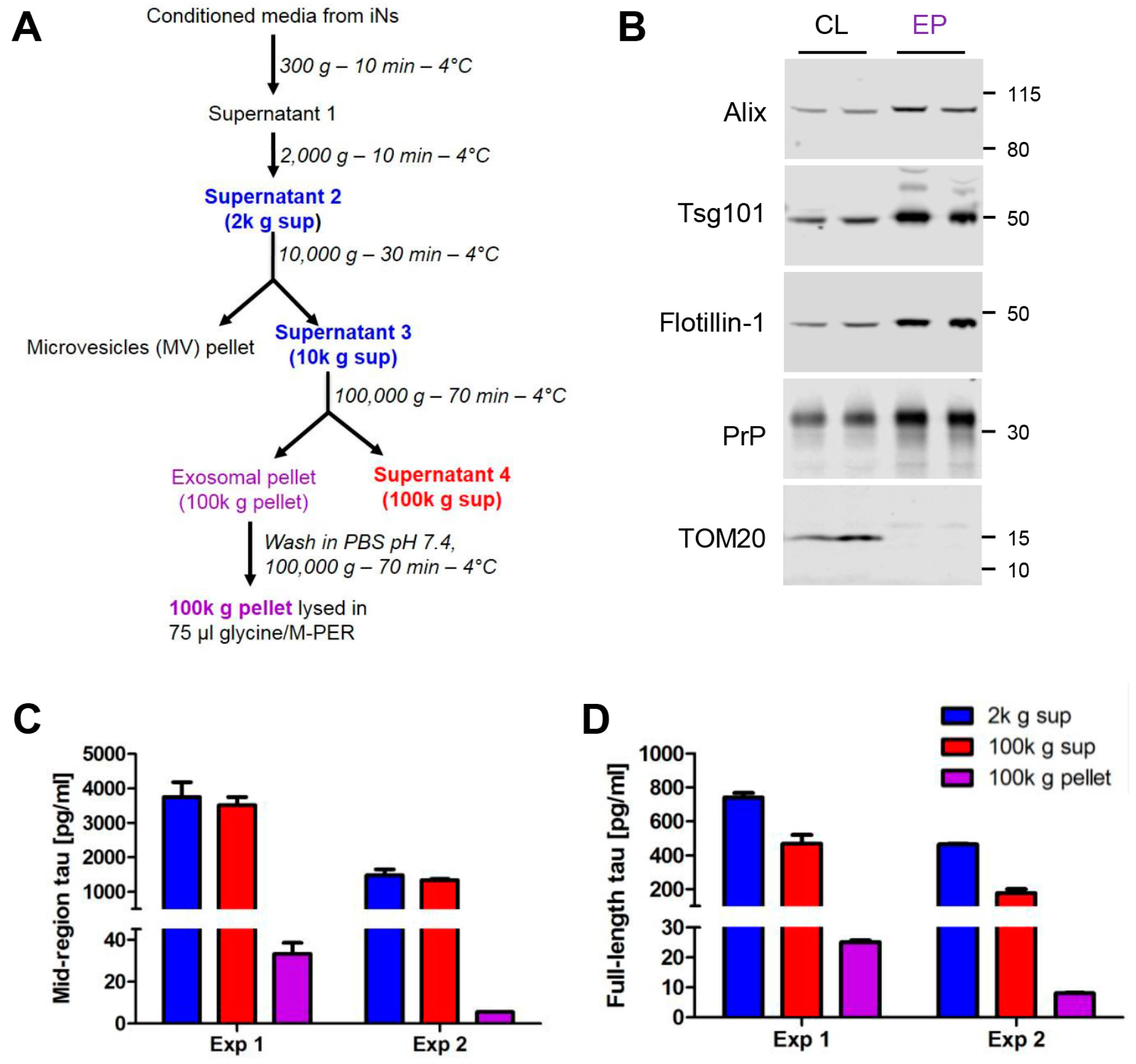
2.1. Human Ipscs-Derived Neurons Release Aggregation-Competent Tau in Exosomes
2.2. Full-Length Tau Is Present in CSF at Low Levels Both Free-Floating and Inside Exosomes
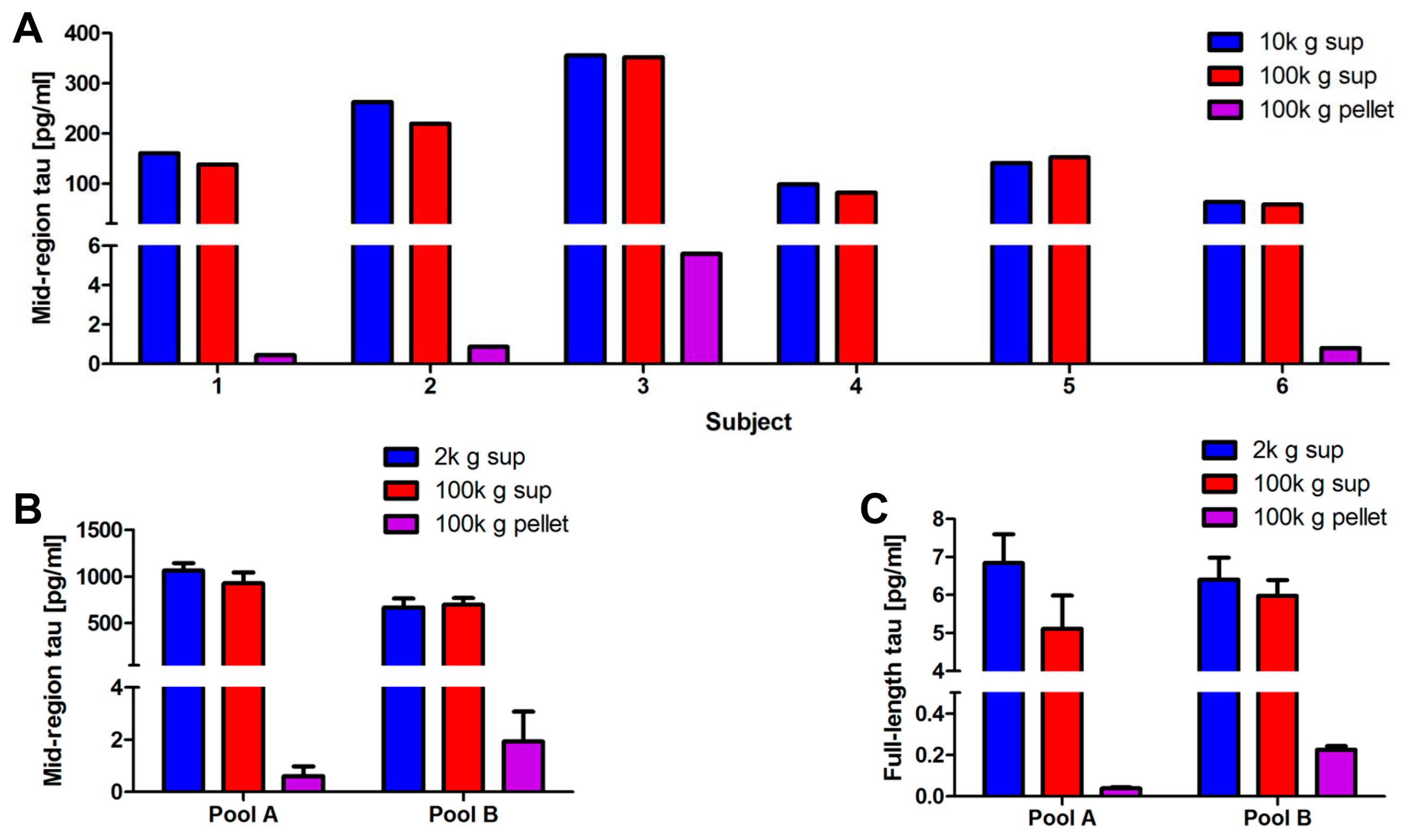
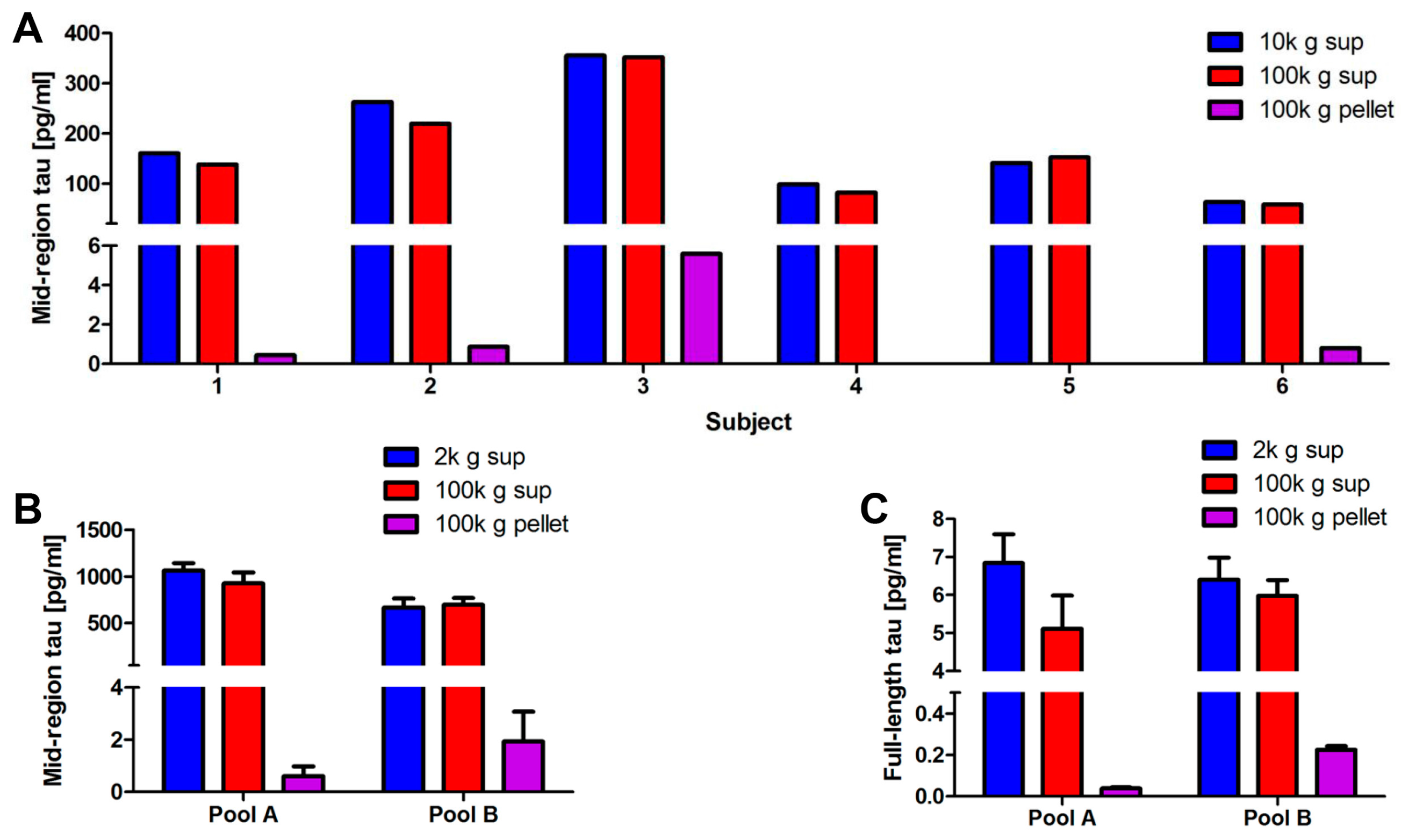
2.3. Optimization of the Method Used to Isolate Neuronal Exosomes from Plasma
2.4. Blood-Derived Neuronal Exosomes Contain Small Amounts of Aggregation-Competent Tau
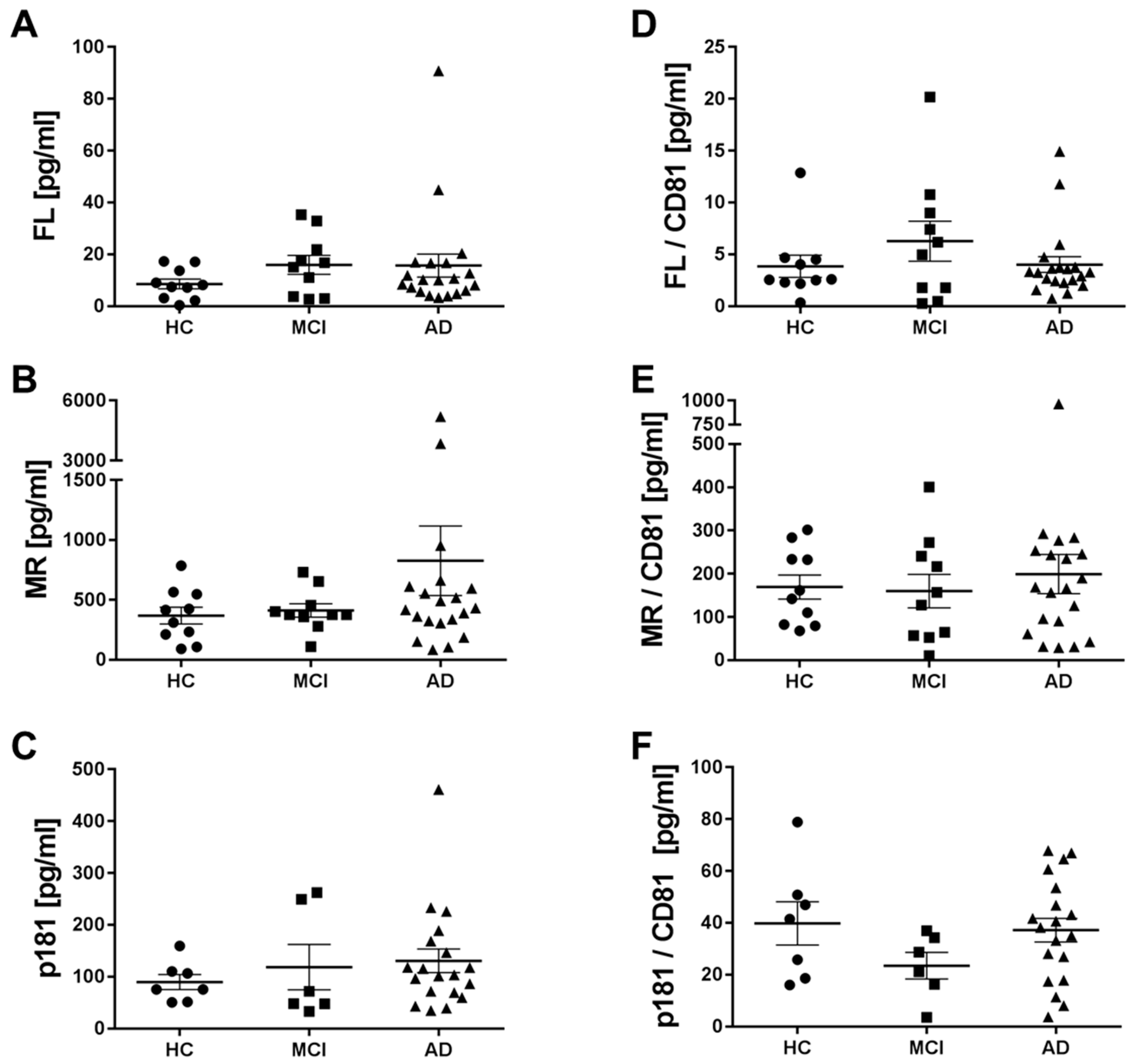
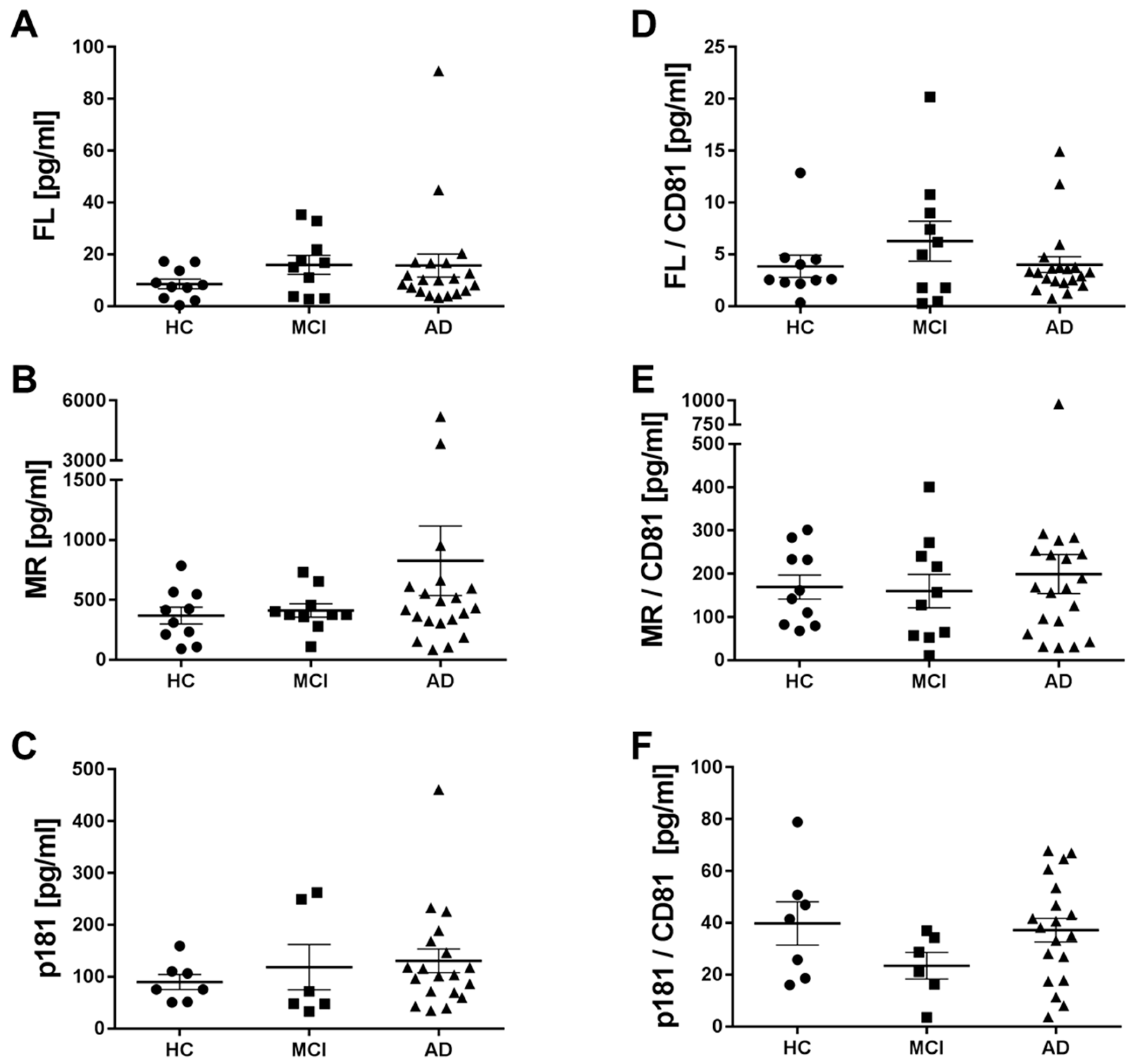
2.5. Levels of Different Types of Tau in Neuronal Exosomes from Healthy Controls, AD and MCI Subjects
3. Discussion
4. Materials and Methods
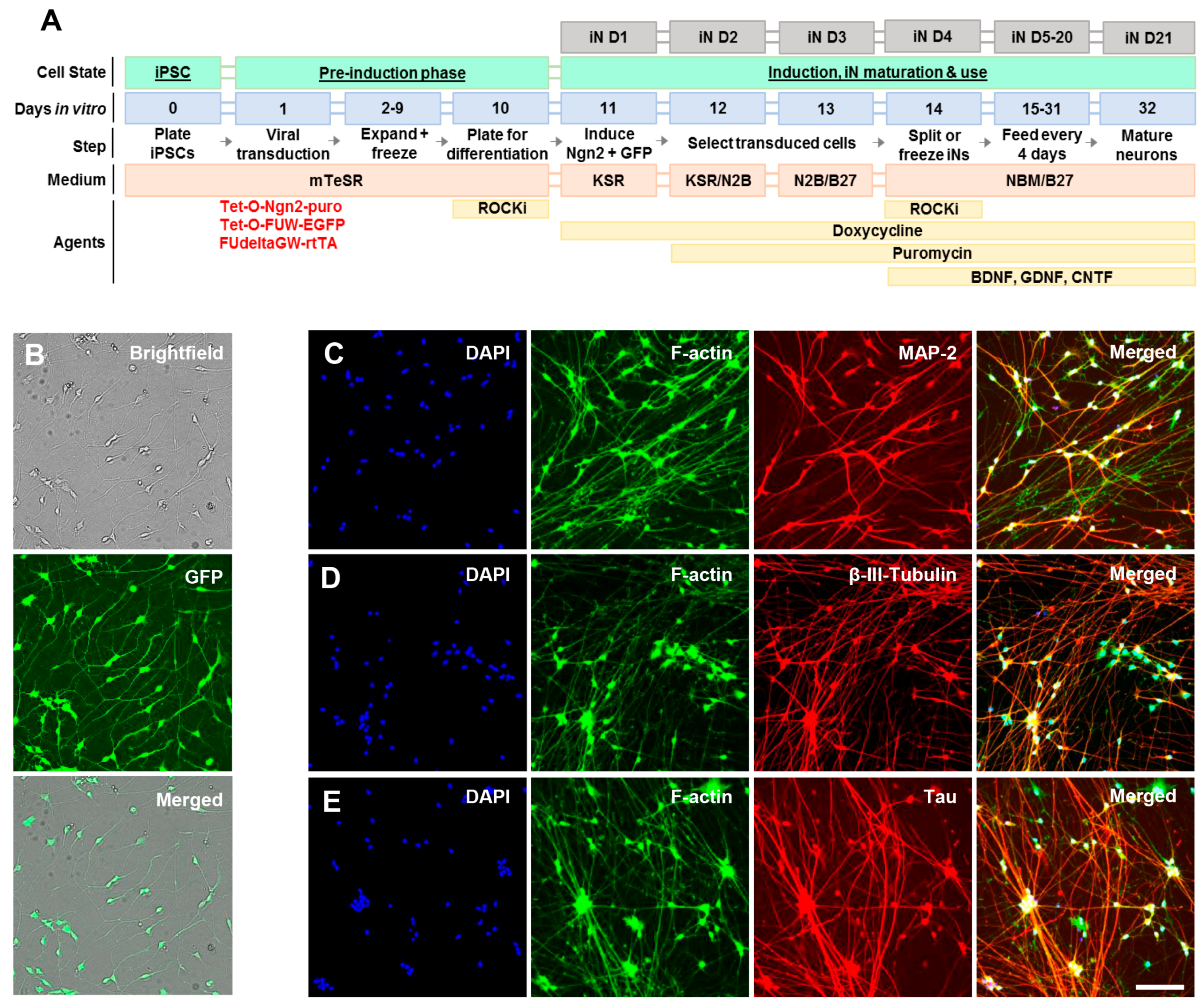
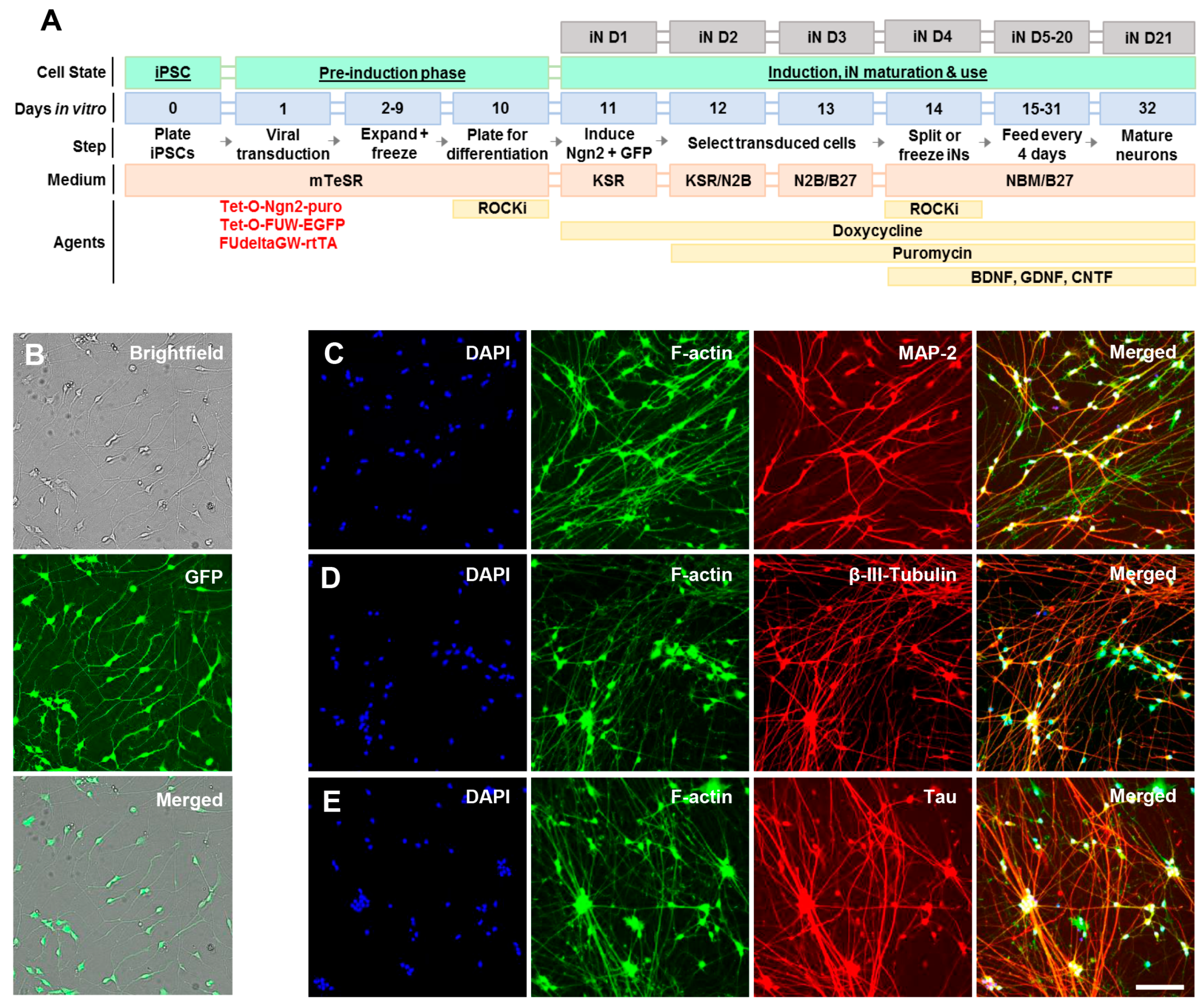
4.1. Generation of Induced Neurons (Ins) from Induced Pluripotent Cells (Ipscs)
4.2. Characterization of Induced Neurons (Ins) by Live Cell Imaging and Immunocytochemistry
4.3. Isolation of Exosomes from in Conditioned Media (CM)
4.4. Western Blotting Analysis of in Cells and Exosomes
4.5. Mid-Region Tau ELISA
4.6. Full-Length Tau ELISA
4.7. Isolation of Exosomes from Human Cerebrospinal Fluid (CSF)
4.8. Plasma Samples and Isolation of Neuronal Exosomes
4.9. CD81 ELISA
4.10. Phosphorylated Threonine-181 (p181) Tau ELISA
4.11. Full-Length Tau Simoa-Based Assay
4.12. Tsg101 MSD Immunoassay
4.13. Characterization of Exosomes by Nanoparticle Tracking Analysis (NanoSight)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β-protein |
| aCSF | Artificial cerebrospinal fluid |
| AD | Alzheimer’s disease |
| Alix | Apoptosis-linked gene 2-interacting protein X |
| ANOVA | Analysis of variance |
| BDNF | Brain derived neurotrophic factor |
| CCDP | Crimson Clinical Discards Program |
| CD81 | Cluster of differentiation 81 |
| CM | Conditioned media |
| CNTF | Ciliary neurotrophic factor |
| CSF | Cerebrospinal fluid |
| DIV | Day in vitro |
| EV | Extracellular vesicle |
| FL | Full-length |
| GDNF | Glial derived neurotrophic factor |
| HBS | Harvard Biomarker Study |
| HC | Healthy control |
| IgG | Immunoglobulin G |
| iN | Induced neuron |
| iPSC | Induced pluripotent stem cell |
| L1CAM | Neural cell adhesion molecule L1 |
| LLoQ | Lower limit of quantitation |
| mAb | Monoclonal antibody |
| MAP-2 | Microtubule-associated protein 2 |
| MCI | Mild cognitive impairment |
| MEM-NEAA | Minimum essential medium with non-essential amino acids |
| MSD | Meso Scale Discovery |
| MTBR | Microtubule binding region |
| MV | Microvesicle |
| MVB | Multivesicular body |
| p181 | Tau phosphorylated at threonine 181 |
| PrP | Prion protein |
| TBST | Tris buffered saline with 0.05% Tween-20 |
| Tsg101 | Tumor susceptibility gene 101 |
| TOM20 | Translocase of the outer membrane-20 |
| ULoQ | Upper limit of quantitation |
References
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Avila, J. The role of extracellular tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 2014, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 2016, 17, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Bjorklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Masliah, E. Alpha-synuclein in lewy body disease and Alzheimer’s disease. Brain Pathol. 1999, 9, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Kanmert, D.; Cantlon, A.; Muratore, C.R.; Jin, M.; O’Malley, T.T.; Lee, G.; Young-Pearse, T.L.; Selkoe, D.J.; Walsh, D.M. C-terminally truncated forms of tau, but not full-length tau or its c-terminal fragments, are released from neurons independently of cell death. J. Neurosci. 2015, 35, 10851–10865. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-beta oligomers induce synaptic damage via tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Begard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buee, L.; Colin, M. Tunneling nanotube (tnt)-mediated neuron-to neuron transfer of pathological tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Raposo, G. Exosomes--vesicular carriers for intercellular communication. Curr. Opin. Cell. Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R. Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol. Direct. 2007, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell. Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev. Cell. Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell. Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation: Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell. Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell. Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Redefining extracellular vesicles. Trends Cell. Biol. 2016, 26, 466–467. [Google Scholar]
- Araldi, E.; Kramer-Albers, E.M.; Hoen, E.N.; Peinado, H.; Psonka-Antonczyk, K.M.; Rao, P.; van Niel, G.; Yanez-Mo, M.; Nazarenko, I. International society for extracellular vesicles: First annual meeting, April 17–21, 2012: Isev-2012. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the international society for extracellular vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Boing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dujardin, S.; Begard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Davies, P.; Mandelkow, E. Tau paired helical filaments from alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 2004, 43, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Goux, W.J.; Kopplin, L.; Nguyen, A.D.; Leak, K.; Rutkofsky, M.; Shanmuganandam, V.D.; Sharma, D.; Inouye, H.; Kirschner, D.A. The formation of straight and twisted filaments from short tau peptides. J. Biol. Chem. 2004, 279, 26868–26875. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Seubert, P.; Cox, T.M.; Motter, R.; Brown, J.P.; Galasko, D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J. Neurochem. 1997, 68, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Meredith, J.E., Jr.; Sankaranarayanan, S.; Guss, V.; Lanzetti, A.J.; Berisha, F.; Neely, R.J.; Slemmon, J.R.; Portelius, E.; Zetterberg, H.; Blennow, K.; et al. Characterization of novel CSF tau and ptau biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e76523. [Google Scholar] [CrossRef] [PubMed]
- Wagshal, D.; Sankaranarayanan, S.; Guss, V.; Hall, T.; Berisha, F.; Lobach, I.; Karydas, A.; Voltarelli, L.; Scherling, C.; Heuer, H.; et al. Divergent CSF tau alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, N.R.; Fenaille, F.; Hirtz, C.; Sergeant, N.; Schraen-Maschke, S.; Vialaret, J.; Buee, L.; Gabelle, A.; Junot, C.; Lehmann, S.; et al. Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J. Proteome Res. 2016, 15, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.X.; et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE 2010, 5, e11853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; O’Nuallain, B.; Hong, W.; Boyd, J.; Lagomarsino, V.N.; O’Malley, T.T.; Liu, W.; Frosch, M.P.; Young-Pearse, T.; Selkoe, D.J.; et al. A new paradigm to assess the therapeutic potential of anti-Aβ antibodies for Alzheimer’s disease. Nat. Commun. 2018. submitted. [Google Scholar]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Stem Cell. Biol. 2006. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Ohrfelt, A.; Portelius, E.; Bjerke, M.; Holtta, M.; Rosen, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Baek, R.; Sondergaard, E.K.; Varming, K.; Jorgensen, M.M. The impact of various preanalytical treatments on the phenotype of small extracellular vesicles in blood analyzed by protein microarray. J. Immunol. Methods 2016, 438, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Kuiperij, H.B.; Verbeek, M.M. Diagnosis of progressive supranuclear palsy: Can measurement of tau forms help? Neurobiol. Aging 2012, 33, e17–e18. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, L.; Darbinian, N.; Goetzl, E.J. Novel window on early human neurodevelopment via fetal exosomes in maternal blood. Ann. Clin. Transl. Neurol. 2016, 3, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Mustapic, M.; Eitan, E.; Werner, J.K., Jr.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma extracellular vesicles enriched for neuronal origin: A potential window into brain pathologic processes. Front. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimers Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.L.; Jicha, G.A.; Shaw, L.M.; Trojanowski, J.Q.; Goetzl, E.J. Plasma neuronal exosomal levels of Alzheimer’s disease biomarkers in normal aging. Ann. Clin. Transl. Neurol. 2016, 3, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of alzheimer’s disease. FASEB J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol 2015, 2, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimers Dement. 2017, 13, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Bright, J.; Hussain, S.; Dang, V.; Wright, S.; Cooper, B.; Byun, T.; Ramos, C.; Singh, A.; Parry, G.; Stagliano, N.; et al. Human secreted tau increases amyloid-beta production. Neurobiol. Aging 2015, 36, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Jeng, A.T.; Goate, A.M. Extracellular tau levels are influenced by variability in tau that is associated with tauopathies. J. Biol. Chem. 2012, 287, 42751–42762. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Jeng, A.T.; Goate, A.M. Calcium phosphatase calcineurin influences tau metabolism. Neurobiol. Aging 2013, 34, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, S.; Hall, G.F. Secretion of human tau fragments resembling CSF-tau in Alzheimer’s disease is modulated by the presence of the exon 2 insert. FEBS Lett. 2010, 584, 3085–3088. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, V.; Mohamed, N.V.; Rivest-McGraw, J.; Bertrand, J.; Lauzon, M.; Leclerc, N. Hyperphosphorylation and cleavage at d421 enhance tau secretion. PLoS ONE 2012, 7, e36873. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; Hall, G.F. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of tau: Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Cadelina, G.W.; Hoque, N.; DeCarr, L.B.; Guss, V.L.; Yang, L.; Sankaranarayanan, S.; Wes, P.D.; Flynn, M.E.; Meredith, J.E.; et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J. Alzheimers Dis. 2011, 24 (Suppl. 2), 127–141. [Google Scholar] [PubMed]
- Reilly, P.; Winston, C.N.; Baron, K.R.; Trejo, M.; Rockenstein, E.M.; Akers, J.C.; Kfoury, N.; Diamond, M.; Masliah, E.; Rissman, R.A.; et al. Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol. Dis. 2017, 106, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Terajima, M.; Miura, M.; Higuchi, S.; Muramatsu, T.; Machida, N.; Seiki, H.; Takase, S.; Clark, C.M.; Lee, V.M.; et al. Tau in cerebrospinal fluid: A potential diagnostic marker in Alzheimer’s disease. Ann. Neurol. 1995, 38, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Malinverno, M.; Gardoni, F.; Alberici, A.; Parnetti, L.; Premi, E.; Bonuccelli, U.; Grassi, M.; Perani, D.; Calabresi, P.; et al. Tau forms in CSF as a reliable biomarker for progressive supranuclear palsy. Neurology 2008, 71, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Iqbal, K.; Bucht, G.; Winblad, B.; Wisniewski, H.M.; Grundke-Iqbal, I. Alzheimer’s disease: Paired helical filament immunoreactivity in cerebrospinal fluid. Acta Neuropathol. 1991, 82, 6–12. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, S.T.; Ardah, M.T.; Johansson, P.; Lomakin, A.; Benedek, G.B.; Roberts, K.A.; Cummins, G.; El Agnaf, O.M.; Svensson, J.; Zetterberg, H.; et al. The ELISA-measured increase in cerebrospinal fluid tau that discriminates Alzheimer’s disease from other neurodegenerative disorders is not attributable to differential recognition of tau assembly forms. J. Alzheimers Dis. 2013, 33, 923–928. [Google Scholar]
- Andreasen, N.; Vanmechelen, E.; Van de Voorde, A.; Davidsson, P.; Hesse, C.; Tarvonen, S.; Raiha, I.; Sourander, L.; Winblad, B.; Blennow, K. Cerebrospinal fluid tau protein as a biochemical marker for Alzheimer’s disease: A community based follow up study. J. Neurol. Neurosurg. Psychiatry 1998, 64, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Ganrot, K.; Laurell, C.B. Measurement of IgG and albumin content of cerebrospinal fluid, and its interpretation. Clin. Chem. 1974, 20, 571–573. [Google Scholar] [PubMed]
- Seyfert, S.; Faulstich, A.; Marx, P. What determines the CSF concentrations of albumin and plasma-derived IgG? J. Neurol. Sci. 2004, 219, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Ohno, H.; Arai, H.; Yamaguchi, H.; Urakami, K.; Park, J.M.; Sato, K.; Kohno, H.; Imahori, K. Phosphorylated tau in human cerebrospinal fluid is a diagnostic marker for Alzheimer’s disease. Neurosci. Lett. 1999, 270, 91–94. [Google Scholar] [CrossRef]
- Keshavan, A.; Mengel, D.; Chen, Z.; Patel, P.; Billinton, A.; Perkinton, M.; Percival-Alwyn, J.; Zetterberg, H.; Johnson, K.; Properzi, M.; et al. Certain plasma N-terminal tau fragments are elevated in AD and AD-MCI compared to controls. In Proceedings of the Alzheimer’s Association International Conference, Chicago, IL, USA, July 2018; pp. 1307–1416. [Google Scholar]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Hampel, H.; Toschi, N.; Baldacci, F.; Zetterberg, H.; Blennow, K.; Kilimann, I.; Teipel, S.J.; Cavedo, E.; Dos Santos, A.M.; Epelbaum, S.; et al. Alzheimer’s disease biomarker-guided diagnostic workflow using the added value of six combined cerebrospinal fluid candidates: Abeta1-42, total-tau, phosphorylated-tau, NFL, neurogranin, and YKL-40. Alzheimers Dement. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC | MCI | Mild AD | Moderate AD | |
|---|---|---|---|---|
| Subjects (n) | 10 | 10 | 10 | 10 |
| Age (Mean ± SD) | 75.90 ± 8.67 | 76.00 ± 7.97 | 75.60 ± 12.89 | 75.00 ± 11.34 |
| MMSE (Mean ± SD) | 29.67 ± 0.52 | 28.10 ± 1.20 | 22.80 ± 6.18 | 15.00 ± 5.24 |
| Sex: | ||||
| Female | 7 | 5 | 4 | 7 |
| Male | 3 | 5 | 6 | 3 |
| APOE allele: | ||||
| ε4/ε4 | - | 1 | 2 | 2 |
| ε4/ε3 | 3 | 2 | 4 | 4 |
| ε4/ε2 | - | 1 | - | - |
| ε3/ε3 | 6 | 5 | 3 | 4 |
| ε3/ε2 | 1 | 1 | 1 | - |
| Diagnosis | CDR | p181 tau (pg/mL) | MR tau (pg/mL) | FL tau (pg/mL) | CD81 A (ng/mL) | CD81 B (ng/mL) | CD81 Avg. (ng/mL) | Tsg101 (ng/mL) | Vesicles/mL (65–305 nm) |
|---|---|---|---|---|---|---|---|---|---|
| HC | 0 | 106.32 | 91.31 | 3.09 | 1.85 | 2.53 | 2.19 | 4.24 * | 7.39 × 1010 |
| HC | 0 | 23.27 * | 212.09 | 9.02 | 3.09 | 3.19 | 3.14 | 6.28 * | 9.30 × 1010 |
| HC | 0 | 110.09 | 564.53 | 17.14 | 12.07 | 10.21 | 11.14 | 25.34 | 6.91 × 1010 |
| HC | 0 | 24.01 * | 106.49 | 17.30 | 1.89 | 2.48 | 2.18 | 17.03 | 7.75 × 1010 |
| HC | 0 | 51.65 | 785.36 | 7.19 | 3.81 | 5.20 | 4.50 | 62.21 | 1.20 × 1011 |
| HC | 0 | 25.21 * | 310.33 | 0.34 * | 1.38 | 1.97 | 1.67 | 8.47 * | 5.88 × 1010 |
| HC | 0 | 75.76 | 415.53 | 7.46 | 3.94 | 5.59 | 4.77 | 13.52 | 4.90 × 1010 |
| HC | 0 | 75.47 | 425.05 | 8.22 | 2.49 | 3.43 | 2.96 | 12.83 | 9.93 × 1010 |
| HC | 0 | 159.50 | 547.89 | 13.67 | 4.81 | 6.23 | 5.52 | 26.26 | 8.64 × 1010 |
| HC | 0 | 50.81 | 232.69 | 2.18 | 1.19 | 2.06 | 1.63 | 10.45 | 1.12 × 1011 |
| MCI | 0.5 | 47.93 | 376.09 | 21.84 | 4.37 | 5.22 | 4.79 | 6.46 * | 1.37 × 1011 |
| MCI | 0.5 | 71.93 | 109.38 | 3.74 | 2.98 | 3.83 | 3.41 | 23.66 | 5.14 × 1010 |
| MCI | 0.5 | 32.98 | 375.45 | 16.79 | 2.34 | 2.73 | 2.54 | 8.07 * | 9.68 × 1010 |
| MCI | 0.5 | 22.35 * | 279.21 | 11.02 | 2.42 | 3.37 | 2.90 | 7.85 * | 7.86 × 1010 |
| MCI | 0.5 | 19.65 * | 653.48 | 32.91 | 2.68 | 2.62 | 2.65 | 20.22 | 9.27 × 1010 |
| MCI | 0.5 | 19.29 * | 455.32 | 15.03 | 2.11 | 3.33 | 2.72 | 5.88 * | 6.73 × 1010 |
| MCI | 0.5 | 47.58 | 358.76 | 2.95 | 2.30 | 3.09 | 2.70 | 10.80 | 8.13 × 1010 |
| MCI | 0.5 | 249.49 | 730.90 | 17.56 | # | 113.64 | 113.64 | 617.83 # | 1.02 × 1011 |
| MCI | 0.5 | 262.32 | 402.86 | 35.30 | 31.68 | 19.31 | 11.54 | 106.61 | 1.37 × 1011 |
| MCI | 0.5 | 38.48 * | 375.07 | 2.74 | 2.07 | 2.47 | 9.50 | 6.00 | 7.34 × 1010 |
| AD | 1 | 103.42 | 518.38 | 10.19 | 5.05 | 5.07 | 5.06 | 14.51 | 5.76 × 1010 |
| AD | 1 | 461.08 | 3850.19 | 90.77 | # | 200.64 | 200.64 | 3203.27 # | 9.62 × 1010 |
| AD | 1 | 69.33 | 390.15 | 3.32 | 2.23 | 3.17 | 2.70 | 8.99 * | 5.49 × 1010 |
| AD | 0.5 | 101.36 | 952.47 | 10.04 | 5.50 | 6.71 | 6.11 | 113.91 | 8.26 × 1010 |
| AD | 2 | 115.47 | 360.99 | 4.80 | 2.39 | 3.79 | 3.09 | 13.05 | 5.97 × 1010 |
| AD | 2 | 233.30 | 152.63 | 16.61 | 7.11 | 9.09 | 8.10 | 30.39 | 8.01 × 1010 |
| AD | 1 | 117.66 | 105.67 | 5.62 | 2.47 | 3.16 | 2.82 | 6.32 * | 5.30 × 1010 |
| AD | 1 | 168.64 | 186.16 | 10.73 | 6.80 | 7.56 | 7.18 | 83.26 | 7.15 × 1010 |
| AD | 1 | 188.85 | 337.34 | 16.71 | 4.71 | 6.75 | 5.73 | 20.00 | 1.21 × 1010 |
| AD | 0.5 | 59.97 | 417.40 | 3.88 | 2.34 | 3.23 | 2.78 | 32.55 | 8.29 × 1010 |
| AD | 2 | 34.95 | 490.67 | 7.25 | 2.36 | 4.13 | 3.25 | 11.66 | 6.81 × 1010 |
| AD | 2 | 146.63 | 663.16 | 8.15 | 3.10 | 4.27 | 3.69 | 16.34 | 1.18 × 1011 |
| AD | 2 | 118.26 | 83.97 | 8.48 | 3.86 | 5.60 | 4.73 | 8.62 * | 1.39 × 1011 |
| AD | 0.5 | 43.28 | 596.59 | 44.91 | 6.58 | 5.80 | 6.19 | 42.78 | 1.09 × 1011 |
| AD | 2 | 39.38 | 612.44 | 6.02 | 8.32 | 7.53 | 7.92 | 29.48 | 8.97 × 1010 |
| AD | 2 | 86.36 | 555.09 | 11.91 | 2.71 | 3.80 | 3.26 | 11.00 | 1.14 × 1011 |
| AD | 1 | 19.47 * | 322.82 | 16.99 | 1.51 | 2.19 | 1.85 | 7.52 * | 8.48 × 1010 |
| AD | 2 | 96.47 | 5198.57 | 20.36 | 8.74 | 8.77 | 8.75 | 81.17 | 1.39 × 1011 |
| AD | 2 | 226.10 | 305.60 | 12.74 | 4.69 | 6.28 | 5.49 | 14.62 | 1.89 × 1011 |
| AD | 2 | 71.86 | 431.69 | 4.05 | 3.42 | 4.90 | 4.16 | 97.17 | 9.51 × 1010 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. https://doi.org/10.3390/ijms19030663
Guix FX, Corbett GT, Cha DJ, Mustapic M, Liu W, Mengel D, Chen Z, Aikawa E, Young-Pearse T, Kapogiannis D, et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. International Journal of Molecular Sciences. 2018; 19(3):663. https://doi.org/10.3390/ijms19030663
Chicago/Turabian StyleGuix, Francesc X., Grant T. Corbett, Diana J. Cha, Maja Mustapic, Wen Liu, David Mengel, Zhicheng Chen, Elena Aikawa, Tracy Young-Pearse, Dimitrios Kapogiannis, and et al. 2018. "Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles" International Journal of Molecular Sciences 19, no. 3: 663. https://doi.org/10.3390/ijms19030663