Transcriptome Analysis Reveals Molecular Signatures of Luteoloside Accumulation in Senescing Leaves of Lonicera macranthoides

Abstract
:1. Introduction
2. Results
2.1. Luteoloside Contents in Different Tissues and Developmental Stages of L. macranthoides
2.2. RNA-Seq Analysis
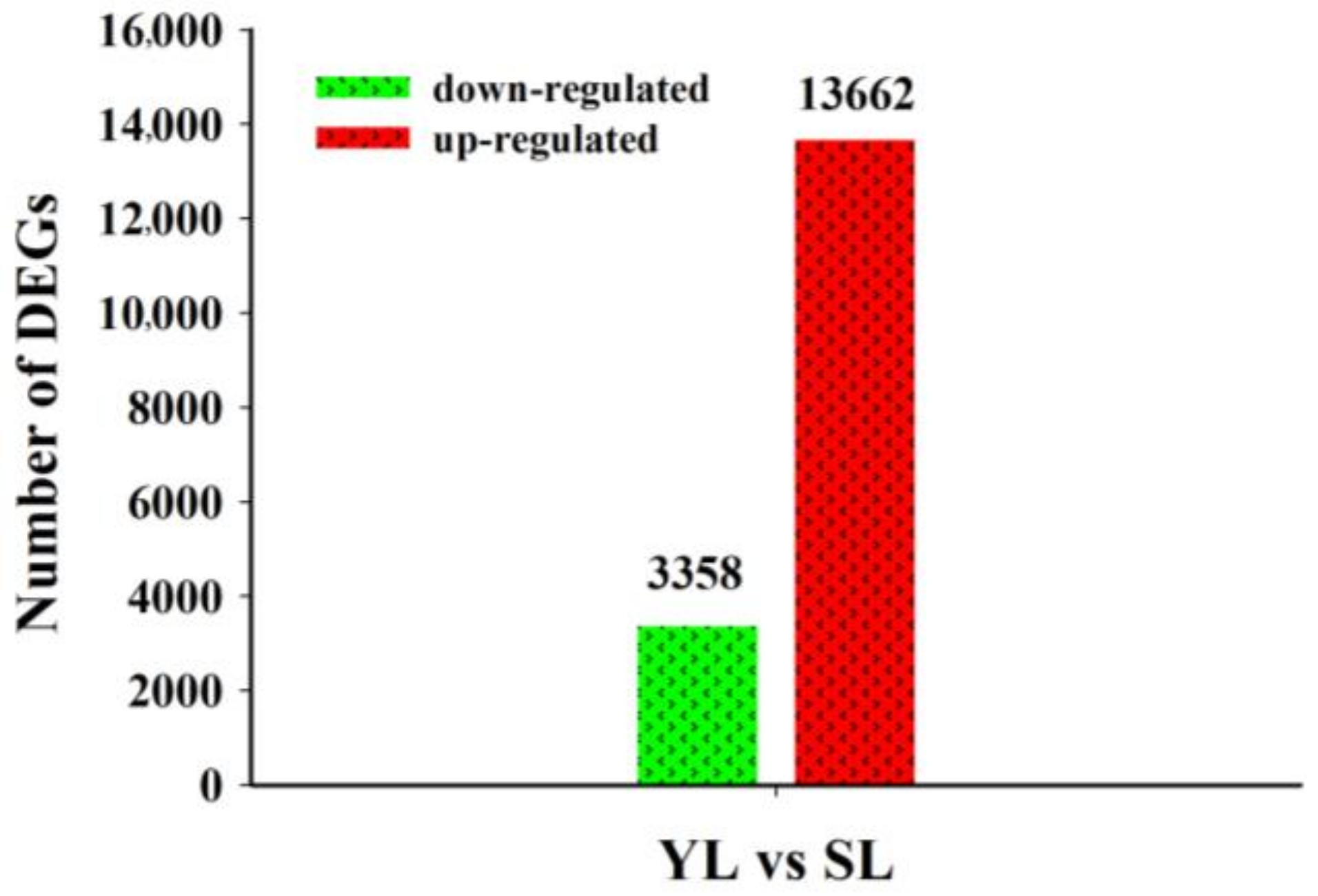
2.3. Differentially Expressed Unigenes (DEGs) in Senescing and Young Leaves
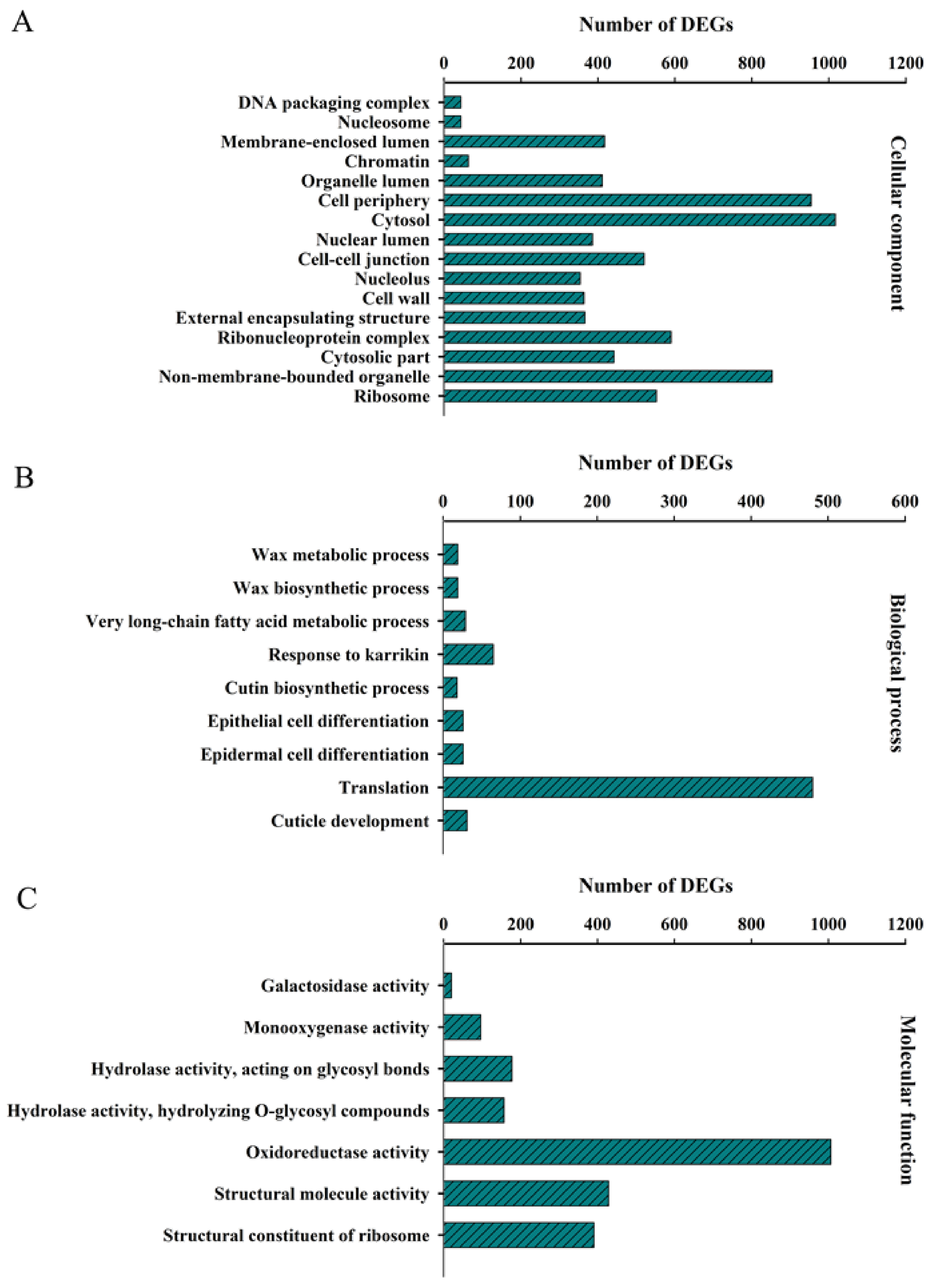
2.4. GO- and KEGG-Based Functional Classification of DEGs
2.5. Identification of Candidate Genes Associated with Luteoloside Biosynthetic Pathways
2.6. Identification of DEGs Involved in MYB, bHLH and WD40 Transcription Factors
3. Discussion
3.1. Molecular Features Underlying Abundant Accumulation of Luteoloside during Leaf Senescence in L. macranthoides
3.2. Transcription Factors as Potential Regulators of Luteoloside Accumulation in Leaves of L. macranthoides
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Luteoloside Contents by High Performance Liquid Chromatography (HPLC)
4.3. RNA Extraction and Illumina Sequencing
4.4. De Novo Transcriptome Assembly and Functional Annotation
4.5. Screening of Differentially Expressed Unigenes
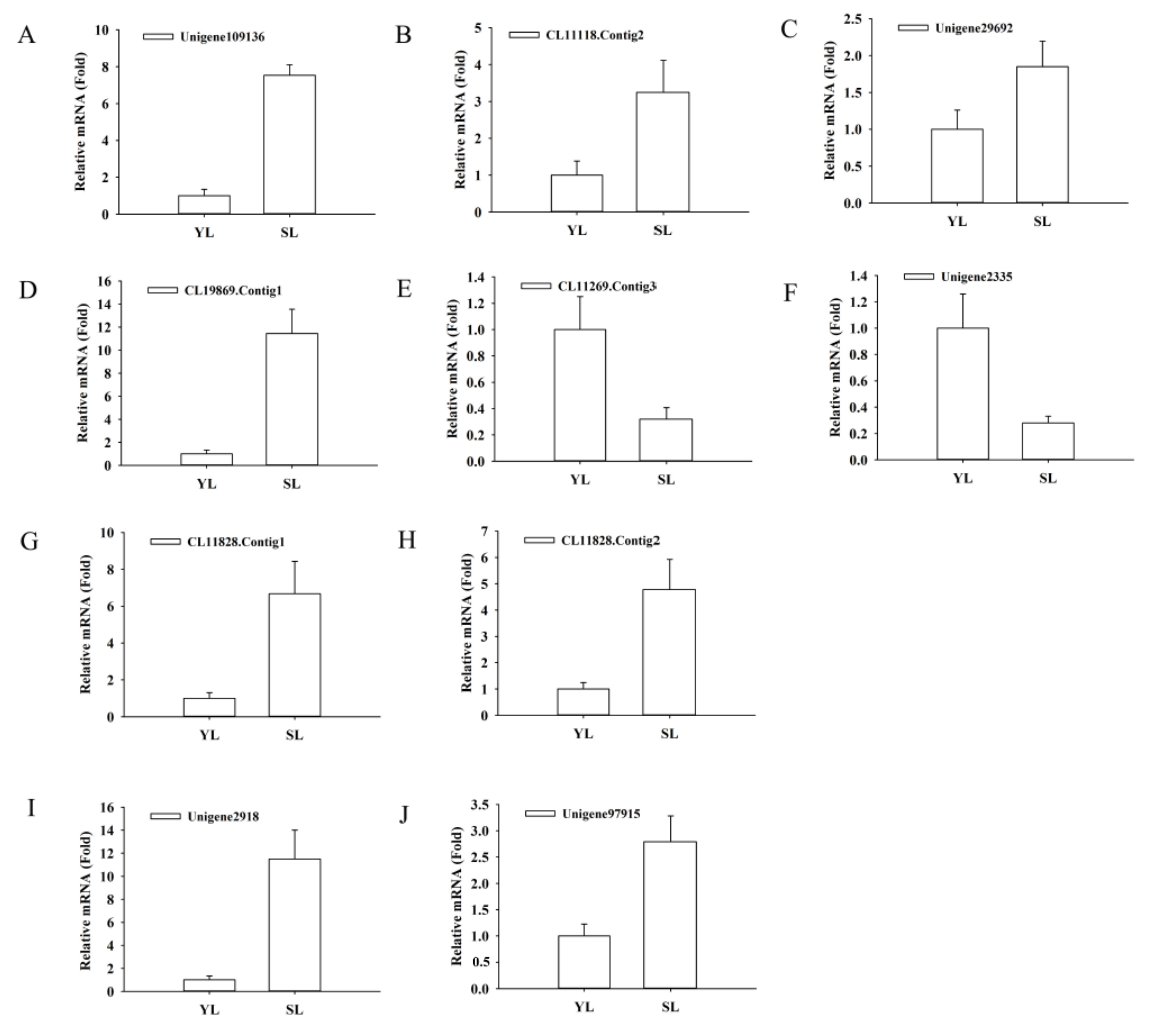
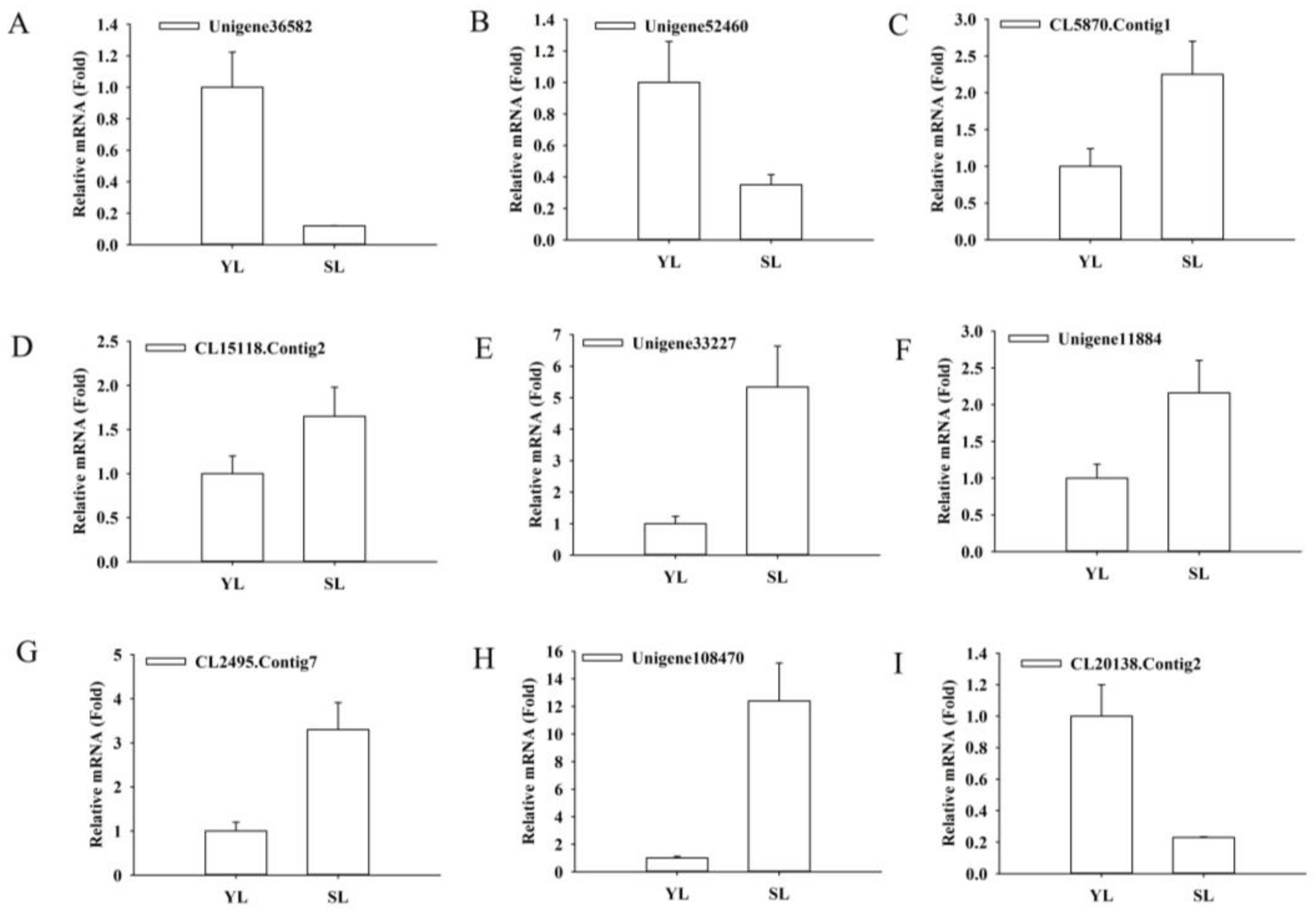
4.6. Validation of RNA-Seq Data by Quantitative Real-Time PCR (qRT-PCR)
4.7. Sequence Deposition
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kong, D.-X.; Li, Y.-Q.; Bai, M.; He, H.-J.; Liang, G.-X.; Wu, H. Correlation between the dynamic accumulation of the main effective components and their associated regulatory enzyme activities at different growth stages in Lonicera japonica Thunb. Ind. Crops Prod. 2017, 96, 16–22. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, G.; Liu, Y.; Xian, Z.; Tang, N. Overexpression of the LmHQT1 gene increases chlorogenic acid production in Lonicera macranthoides Hand-Mazz. Acta Physiol. Plant. 2017, 39, 27. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Li, P. Capillary high-performance liquid chromatography with mass spectrometry for simultaneous determination of major flavonoids, iridoid glucosides and saponins in Flos Lonicerae. J. Chromatogr. A 2007, 1157, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.L.; Li, P.; Song, Y.; Chai, X.Y.; Ma, D.Y. Qualitative and quantitative analysis of active flavonoids in Flos Lonicerae by capillary zone electrophoresis coupled with solid-phase extraction. J. Sep. Sci. 2005, 28, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Pan, H.; Li, M.; Miao, X.; Ding, H. Lonicera japonica Thunb.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2011, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.Z.; Qi, Q.; Tao, L.; Zhao, Q.; Mu, R.; Gu, H.Y.; Wang, M.; Feng, X.; Guo, Q.L. Macranthoside B, a hederagenin saponin extracted from Lonicera macranthoides and its anti-tumor activities in vitro and in vivo. Food Chem. Toxicol. 2009, 47, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, X.-C.; Liu, Y.; Du, H.; Shu, Q.-Y.; Su, S.; Wang, L.-J.; Li, S.-S.; Wang, L.-S. Flavone synthases from Lonicera japonica and L. macranthoides reveal differential flavone accumulation. Sci. Rep. 2016, 6, 19245. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Z.Y.; Jiang, C.; Wang, X.M.; Huang, L.Q. Exploiting genes and functional diversity of chlorogenic acid and luteolin biosyntheses in Lonicera japonica and their substitutes. Gene 2014, 534, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Li, Z.; He, L.; Wang, D. HPLC-ESI-MS/MS analysis and pharmacokinetics of luteoloside, a potential anticarcinogenic component isolated from Lonicera japonica, in beagle dogs. Biomed. Chromatogr. 2013, 27, 311–317. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xu, X.; Li, Y.; Li, C.; Zhu, Y.; Yan, H.; Sun, Z.; Sun, C.; Song, J.; Bi, Y.A.; et al. Transcriptome Analysis of Buds and Leaves Using 454 Pyrosequencing to Discover Genes Associated with the Biosynthesis of Active Ingredients in Lonicera japonica Thunb. PLoS ONE 2013, 8, e62922. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tang, N.; You, Y.; Lan, J.; Liu, Y.; Li, Z. Transcriptome Analysis Reveals the Mechanism Underlying the Production of a High Quantity of Chlorogenic Acid in Young Leaves of Lonicera macranthoides Hand.-Mazz. PLoS ONE 2015, 10, e0137212. [Google Scholar] [CrossRef] [PubMed]
- Kitada, C.; Gong, Z.; Tanaka, Y.; Yamazaki, M.; Saito, K. Differential expression of two cytochrome P450s involved in the biosynthesis of flavones and anthocyanins in chemo-varietal forms of Perilla frutescens. Plant Cell Physiol. 2001, 42, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Song, L.; Li, M.; Liu, G.; Chu, Y.; Ma, L.; Zhou, Y.; Wang, X.; Gao, W.; Qin, S.; et al. Genetic variation and metabolic pathway intricacy govern the active compound content and quality of the Chinese medicinal plant Lonicera japonica thunb. BMC Genom. 2012, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Kamochi, H.; Suzuki, H.; Nakamura, M.; Takahashi, H.; Hatada, T.; Saito, K.; Yamazaki, M. De novo transcriptome assembly and characterization of nine tissues of Lonicera japonica to identify potential candidate genes involved in chlorogenic acid, luteolosides, and secoiridoid biosynthesis pathways. J. Nat. Med. 2017, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mehrtens, F.; Kranz, H.; Bednarek, P.; Weisshaar, B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Ishihara, H.; Barsch, G.H.A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Czemmel, S.; Stracke, R.; Weisshaar, B.; Cordon, N.; Harris, N.N.; Walker, A.R.; Robinson, S.P.; Bogs, J. The Grapevine R2R3-MYB Transcription Factor VvMYBF1 Regulates Flavonol Synthesis in Developing Grape Berries. Plant Physiol. 2009, 151, 1513–1530. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Khaidun, A.B.M.; Chen, J.; Zhang, C.; Lv, H.; Yuan, L.; Wang, Y. A R2R3-MYB Transcription Factor Regulates the Flavonol Biosynthetic Pathway in a Traditional Chinese Medicinal Plant, Epimedium sagittatum. Front. Plant Sci. 2016, 7, 1089. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Honda, C.; Hatsuyama, Y.; Igarashi, M.; Bessho, H.; Moriguchi, T. Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol. 2007, 48, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Takos, A.M.; Jaffe, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ishimaru, M.; Hiraoka, K.; Honda, C. MYB-related genes of the Kyoho grape (Vitis labruscana) regulate anthocyanin biosynthesis. Planta 2002, 215, 924–933. [Google Scholar] [PubMed]
- Walker, A.R.; Lee, E.; Bogs, J.; McDavid, D.A.J.; Thomas, M.R.; Robinson, S.P. White grapes arose through the mutation of two similar and adjacent regulatory genes. Plant J. 2007, 49, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.Q.; Wang, Y.L.; Yang, S.; Xu, Y.T.; Chen, X.S. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta 2010, 232, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Le Gourrierec, J.; Baudry, A.; Huep, G.; Lanet, E.; Debeaujon, I.; Routaboul, J.M.; Alboresi, A.; Weisshaar, B.; Lepiniec, L. MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. 2008, 55, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.L.; Cominelli, E.; Bailey, P.; Parr, A.; Mehrtens, F.; Jones, J.; Tonelli, C.; Weisshaar, B.; Martin, C. Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. Embo J. 2000, 19, 6150–6161. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, J.B.; Cho, K.J.; Cheon, C.I.; Sung, M.K.; Choung, M.G.; Roh, K.H. Arabidopsis R2R3-MYB transcription factor AtMYB60 functions as a transcriptional repressor of anthocyanin biosynthesis in lettuce (Lactuca sativa). Plant Cell Rep. 2008, 27, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Tamagnone, L.; Merida, A.; Parr, A.; Mackay, S.; Culianez-Macia, F.A.; Roberts, K.; Martin, C. The AmMYB308 and AmMYB330 transcription factors from antirrhinum regulate phenylpropanoid and lignin biosynthesis in transgenic tobacco. Plant Cell 1998, 10, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; de Vos, C.H.R.; Wein, M.; Sun, Z.K.; Greco, R.; Kroon, A.; Mol, J.N.M.; O’Connell, A.P. The strawberry FaMYB1 transcription factor suppresses anthocyanin and flavonol accumulation in transgenic tobacco. Plant J. 2001, 28, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Li, S. Transcriptional control of flavonoid biosynthesis: Fine-tuning of the MYB-bHLH-WD40 (MBW) complex. Plant Signal. Behav. 2014, 9, e27522. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.-W.; Liu, P.; Wang, Y.; Qin, D.; Zhao, L.-J. De novo transcriptome sequencing of blue honeysuckle fruit (Lonicera caerulea L.) and analysis of major genes involved in anthocyanin biosynthesis. Acta Physiol. Plant. 2016, 38, 180. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Quan, Q.; Zhou, X.; Zhu, Y.; Lan, Y.; Li, S.; Yu, Y.; Cheng, Z. A comparative study of Lonicera japonica with related species: Morphological characteristics, ITS sequences and active compounds. Biochem. Syst. Ecol. 2014, 54, 198–207. [Google Scholar] [CrossRef]
- Shi, R.; Sun, Y.H.; Li, Q.Z.; Heber, S.; Sederoff, R.; Chiang, V.L. Towards a Systems Approach for Lignin Biosynthesis in Populus trichocarpa: Transcript Abundance and Specificity of the Monolignol Biosynthetic Genes. Plant Cell Physiol. 2010, 51, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, M.J.; Lee, J.Y.; Kim, J.K.; Ha, S.-H.; Lim, S.-H. Molecular and Biochemical Analysis of Two Rice Flavonoid 3′-Hydroxylase to Evaluate Their Roles in Flavonoid Biosynthesis in Rice Grain. Int. J. Mol. Sci. 2016, 17, 1549. [Google Scholar] [CrossRef] [PubMed]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Pandey, A.; Tiwari, M.; Chandrashekar, K.; Sidhu, O.P.; Asif, M.H.; Chakrabarty, D.; Singh, P.K.; Trivedi, P.K.; Nath, P.; et al. Modulation of transcriptome and metabolome of tobacco by Arabidopsis transcription factor, AtMYB12, leads to insect resistance. Plant Physiol. 2010, 152, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Misra, P.; Trivedi, P.K. Constitutive expression of Arabidopsis MYB transcription factor, AtMYB11, in tobacco modulates flavonoid biosynthesis in favor of flavonol accumulation. Plant Cell Rep. 2015, 34, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Jahns, O.; Keck, M.; Tohge, T.; Niehaus, K.; Fernie, A.R.; Weisshaar, B. Analysis of PRODUCTION OF FLAVONOL GLYCOSIDES-dependent flavonol glycoside accumulation in Arabidopsis thaliana plants reveals MYB11-, MYB12-and MYB111-independent flavonol glycoside accumulation. New Phytol. 2010, 188, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Adato, A.; Mandel, T.; Mintz-Oron, S.; Venger, I.; Levy, D.; Yativ, M.; Dominguez, E.; Wang, Z.; de Vos, R.C.H.; Jetter, R.; et al. Fruit-Surface Flavonoid Accumulation in Tomato is Controlled by a SIMYB12-Regulated Transcriptional Network. PLoS Genet. 2009, 5, e1000777. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Pireyre, M.; Burow, M. Regulation of MYB and bHLH Transcription Factors: A Glance at the Protein Level. Mol. Plant 2015, 8, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.J.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.J.; Grain, D.; Bobet, S.; Le Gourrierec, J.; Thevenin, J.; Kelemen, Z.; Lepiniec, L.; Dubos, C. Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 2014, 202, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, L.W.; Zhou, X.J.; Burke, S.; Wu, X.L.; Prior, R.L.; Li, L. The Purple Cauliflower Arises from Activation of a MYB Transcription Factor. Plant Physiol. 2010, 154, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Cho, M.; Choi, M.G.; Das, P.K.; Lee, S.K.; Choi, S.B.; Park, Y.I. Identification of genes that may regulate the expression of the transcription factor production of anthocyanin pigment 1 (PAP1)/MYB75 involved in Arabidopsis anthocyanin biosynthesis. Plant Cell Rep. 2015, 34, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Quattrocchio, F.; Wing, J.F.; Leppen, H.T.C.; Mol, J.N.M.; Koes, R.E. Regulatory Genes-Controlling Anthocyanin Pigmentation Are Functionally Conserved among Plant-Species and Have Distinct Sets of Target Genes. Plant Cell 1993, 5, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Number | Total Length (nt) | Mean Length (nt) | N50 | Distinct Clusters | Distinct Singletons |
|---|---|---|---|---|---|---|
| SL-Trinity | 260,079 | 202,070,131 | 777 | 1238 | - | - |
| YL-Trinity | 173,214 | 165,117,523 | 953 | 1607 | - | - |
| SL-Unigene | 183,667 | 165,907,666 | 903 | 1501 | 57,995 | 125,672 |
| YL-Unigene | 122,824 | 138,943,927 | 1131 | 1859 | 48,507 | 74,317 |
| All-Unigene | 158,842 | 171,974,902 | 1083 | 1743 | 57,337 | 101,505 |
| EC Number | Accession No. | Reads Mean YL | Reads Mean SL | log2Ratio (SL/YL) | Annotation |
|---|---|---|---|---|---|
| EC:4.3.1.24 | Unigene109136 | 0.5 | 66 | 7.44 | Solanum tuberosum PAL2 |
| Unigene27450 | 2273.5 | 394.5 | −2.21 | Lonicera japonica PAL3 | |
| Unigene29003 | 904 | 89.5 | −3.01 | Lonicera japonica PAL2 | |
| EC:1.14.13.11 | CL11118.Contig2 | 145 | 1117 | 3.3 | Lonicera japonica C4H |
| EC:6.2.1.12 | CL15146.Contig1 | 0 | 40 | Inf | Arabidopsis thaliana 4CLL7 |
| CL7954.Contig3 | 1612 | 138 | −3.22 | Arabidopsis thaliana 4CLL10 | |
| Unigene100595 | 0 | 69.5 | Inf | Petroselinum crispum 4CL2 | |
| Unigene141050 | 0 | 165.5 | Inf | Arabidopsis thaliana 4CLL10 | |
| Unigene26593 | 1950 | 4922.5 | 1.67 | Arabidopsis thaliana 4CLL7 | |
| Unigene29692 | 1735.5 | 4766 | 1.79 | Arabidopsis thaliana 4CLL7 | |
| Unigene66358 | 0 | 29.5 | Inf | Oryza sativa 4CL5 | |
| Unigene73587 | 0.5 | 155 | 8.67 | Petroselinum crispum 4CL2 | |
| Unigene75066 | 0 | 49 | Inf | Arabidopsis thaliana 4CL4 | |
| Unigene80421 | 0 | 49 | Inf | Nicotiana tabacum 4CL2 | |
| Unigene81606 | 0 | 124 | Inf | Arabidopsis thaliana 4CLL10 | |
| Unigene82586 | 0.5 | 154 | 8.67 | Arabidopsis thaliana 4CLL1 | |
| Unigene84369 | 0 | 34.5 | Inf | Arabidopsis thaliana 4CLL10 | |
| Unigene99638 | 0 | 181.5 | Inf | Arabidopsis thaliana 4CLL7 | |
| EC:2.3.1.74 | CL11967.Contig1 | 5886 | 28.5 | −7.34 | Lonicera japonica CHS |
| CL19869.Contig1 | 0.5 | 59 | 7.273 | Hordeum vulgare CHS2 | |
| Unigene12731 | 236 | 7 | −4.74 | Lonicera macranthoides CHS | |
| Unigene1352 | 5294 | 15.5 | −8.06 | Lonicera japonica CHS | |
| Unigene23683 | 31,797 | 852.5 | −4.89 | Lonicera hypoglauca CHS2 | |
| Unigene23684 | 12,441.5 | 400 | −4.63 | Lonicera japonica CHS2 | |
| Unigene40151 | 416 | 1 | −8.35 | Lonicera japonica CHS | |
| Unigene69273 | 1291 | 35 | −4.88 | Lonicera japonica CHS2 | |
| EC:5.5.1.6 | CL11269.Contig3 | 1217.5 | 110.5 | −3.13 | Lonicera japonica CHI2 |
| CL16735.Contig2 | 4566 | 377.5 | −3.27 | Lonicera japonica CHI1 | |
| EC:1.14.11.22 | Unigene2335 | 6778 | 781 | −2.77 | Lonicera macranthoidesFNS II |
| EC:1.14.13.21 | CL11828.Contig1 | 1840 | 26,424.5 | 4.2 | Petunia hybrida F3′H |
| CL11828.Contig2 | 13.5 | 196.5 | 4.25 | Arabidopsis thaliana F3′H | |
| CL7653.Contig1 | 0 | 31.5 | Inf | Solanum melongena F3′H | |
| Unigene102655 | 0 | 56 | Inf | Catharanthus roseus F3′H | |
| Unigene3751 | 799.5 | 21 | −4.9 | Petunia hybrida F3′H | |
| Unigene65437 | 0 | 44 | Inf | Arabidopsis thaliana F3′H | |
| Unigene76746 | 0 | 24 | Inf | Zea mays F3′H | |
| Unigene18958 | 38,738 | 1897 | −4 | Petunia hybrida F3′H | |
| EC:2.4.1.81 | CL5848.Contig3 | 1370 | 161 | −2.8 | Fragaria ananassa UFGT6 |
| CL8885.Contig1 | 1477 | 37.5 | −5 | Fragaria ananassa UFGT6 | |
| CL8885.Contig2 | 1725 | 27 | −5.7 | Fragaria ananassa UFGT6 | |
| CL8885.Contig3 | 707 | 3 | −7.6 | Manihot esculenta GT2 | |
| Unigene2918 | 12 | 279.5 | 4.82 | Fragaria ananassa UFGT6 | |
| Unigene4567 | 208 | 3 | −5.8 | Fragaria ananassa UFGT7 | |
| Unigene97915 | 5.5 | 114 | 4.69 | Fragaria ananassa UFGT6 |
| Accession No. | Reads Mean YL | Reads Mean SL | log2Ratio (SL/YL) | Annotation |
|---|---|---|---|---|
| MYB | ||||
| CL11844.Contig2 | 363.5 | 35 | −3.04 | Paeonia suffruticosa MYB1 |
| CL13949.Contig1 | 147.5 | 1.5 | −6.27 | Vitis vinifera AS1 |
| CL13949.Contig2 | 88 | 0.5 | −7.2 | Vitis vinifera AS1 |
| CL13949.Contig3 | 466 | 10 | −5.22 | Vitis vinifera AS1 |
| CL14249.Contig3 | 267.5 | 1414 | 2.70 | Gossypium hirsutum GhMYB9 |
| CL14844.Contig2 | 39.5 | 0 | −Inf | Vitis vinifera MYB39 |
| CL15118.Contig2 | 3419.5 | 8646.5 | 1.67 | Vitis vinifera MYB1R1 |
| CL20138.Contig1 | 205.5 | 19 | −3.11 | Arabidopsis thaliana TT2 |
| CL20138.Contig2 | 700.5 | 42 | −3.75 | Arabidopsis thaliana MYB5 |
| CL2957.Contig1 | 0 | 163.5 | Inf | Solanum tuberosum MYB1R1 |
| CL3616.Contig1 | 2.5 | 258.5 | 7.05 | Arabidopsis thaliana MYB48 |
| CL3616.Contig3 | 35 | 1308.5 | 5.51 | Arabidopsis thaliana MYB48 |
| CL3616.Contig5 | 10 | 197.5 | 4.58 | Arabidopsis thaliana MYB48 |
| CL4609.Contig1 | 124.5 | 528.5 | 2.43 | Theobroma cacao myb2 |
| CL5870.Contig1 | 483.5 | 1731.5 | 2.21 | Vitis vinifera MYB75 |
| CL5870.Contig2 | 191 | 711.5 | 2.27 | Vitis vinifera MYB75 |
| CL6351.Contig2 | 120 | 9 | −3.43 | Theobroma cacao Myb 106 |
| CL6351.Contig3 | 229.5 | 8.5 | −4.4 | Theobroma cacao Myb 106 |
| CL8018.Contig2 | 5 | 162 | 5.37 | Antirrhinum majus MYB340 |
| CL8018.Contig4 | 232 | 6696 | 5.18 | Antirrhinum majus MYB305 |
| Unigene20238 | 3419 | 8733.5 | 1.71 | Epimedium sagittatum MYB13 |
| Unigene23056 | 86 | 360 | 2.42 | Petunia hybrida ODO1 |
| Unigene32379 | 1639 | 358.5 | −1.88 | Vitis vinifera MYB44 |
| Unigene36582 | 901.5 | 0.5 | −10.4 | Arabidopsis thaliana MYB12 |
| Unigene37949 | 59.5 | 0.5 | −6.48 | Arabidopsis thaliana MYB46 |
| Unigene42805 | 45.5 | 0.5 | −6.12 | Vitis vinifera MYB6 |
| Unigene43758 | 138 | 4 | −4.85 | Vitis vinifera myb306 |
| Unigene47067 | 63.5 | 0 | -Inf | Solanum lycopersicum MYB46 |
| Unigene52460 | 132 | 0 | −Inf | Arabidopsis thaliana MYB12 |
| Unigene70407 | 1.5 | 61 | 5.68 | Oryza sativa MYB4 |
| bHLH | ||||
| CL10785.Contig3 | 243.5 | 0.5 | −8.66 | Arabidopsis thaliana bHLH93 |
| CL10785.Contig6 | 107.5 | 0 | −Inf | Arabidopsis thaliana bHLH93 |
| CL10785.Contig7 | 199.5 | 1 | −7.32 | Arabidopsis thaliana bHLH93 |
| CL11820.Contig21 | 238.5 | 6.5 | −4.85 | Arabidopsis thaliana bHLH4 |
| CL11820.Contig6 | 119 | 4.5 | −4.38 | Arabidopsis thaliana bHLH4 |
| CL12435.Contig2 | 327 | 37 | −2.81 | Arabidopsis thaliana bHLH74 |
| CL18193.Contig2 | 97 | 0 | −Inf | Arabidopsis thaliana bHLH25 |
| CL2495.Contig10 | 37.5 | 317 | 3.42 | Arabidopsis thaliana bHLH78 |
| CL2495.Contig3 | 82 | 904.5 | 3.79 | Arabidopsis thaliana bHLH78 |
| CL2495.Contig7 | 25.5 | 218 | 3.42 | Arabidopsis thaliana bHLH78 |
| CL2495.Contig9 | 10.5 | 111 | 3.72 | Arabidopsis thaliana bHLH78 |
| CL3128.Contig3 | 384.5 | 1947 | 2.7 | Arabidopsis thaliana bHLH47 |
| CL3403.Contig4 | 642 | 94.5 | −2.42 | Arabidopsis thaliana bHLH93 |
| CL3655.Contig4 | 8.5 | 99.5 | 3.93 | Arabidopsis thaliana bHLH113 |
| CL4290.Contig2 | 245.5 | 870.5 | 2.18 | Arabidopsis thaliana bHLH143 |
| CL4720.Contig6 | 37 | 211.5 | 2.88 | Arabidopsis thaliana bHLH123 |
| CL661.Contig4 | 20.5 | 231 | 3.88 | Arabidopsis thaliana bHLH25 |
| CL7365.Contig10 | 145.5 | 467 | 2.02 | Arabidopsis thaliana bHLH66 |
| CL8229.Contig1 | 48 | 0 | −Inf | Arabidopsis thaliana bHLH135 |
| CL8229.Contig2 | 38.5 | 0 | −Inf | Arabidopsis thaliana bHLH135 |
| Unigene11884 | 22 | 106 | 2.65 | Arabidopsis thaliana bHLH113 |
| Unigene19084 | 1634 | 43.5 | −4.9 | Arabidopsis thaliana bHLH117 |
| Unigene2295 | 412.5 | 53.5 | −2.61 | Vitis vinifera bHLH74 |
| Unigene23034 | 81 | 351.5 | 2.47 | Arabidopsis thaliana bHLH147 |
| Unigene29099 | 650.5 | 112 | −2.21 | Arabidopsis thaliana bHLH79 |
| Unigene33227 | 27 | 427 | 4.37 | Arabidopsis thaliana bHLH113 |
| Unigene37456 | 135.5 | 1 | −6.77 | Arabidopsis thaliana bHLH71 |
| Unigene48118 | 139.5 | 1 | −6.83 | Arabidopsis thaliana bHLH14 |
| Unigene6423 | 312.5 | 54.5 | −2.2 | Arabidopsis thaliana bHLH79 |
| Unigene77068 | 0.5 | 49 | 7.02 | Arabidopsis thaliana bHLH49 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Liu, G.; Tang, N.; Li, Z. Transcriptome Analysis Reveals Molecular Signatures of Luteoloside Accumulation in Senescing Leaves of Lonicera macranthoides. Int. J. Mol. Sci. 2018, 19, 1012. https://doi.org/10.3390/ijms19041012
Chen Z, Liu G, Tang N, Li Z. Transcriptome Analysis Reveals Molecular Signatures of Luteoloside Accumulation in Senescing Leaves of Lonicera macranthoides. International Journal of Molecular Sciences. 2018; 19(4):1012. https://doi.org/10.3390/ijms19041012
Chicago/Turabian StyleChen, Zexiong, Guohua Liu, Ning Tang, and Zhengguo Li. 2018. "Transcriptome Analysis Reveals Molecular Signatures of Luteoloside Accumulation in Senescing Leaves of Lonicera macranthoides" International Journal of Molecular Sciences 19, no. 4: 1012. https://doi.org/10.3390/ijms19041012