PCLPred: A Bioinformatics Method for Predicting Protein–Protein Interactions by Combining Relevance Vector Machine Model with Low-Rank Matrix Approximation

Abstract
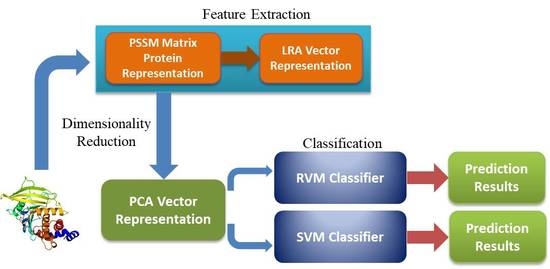
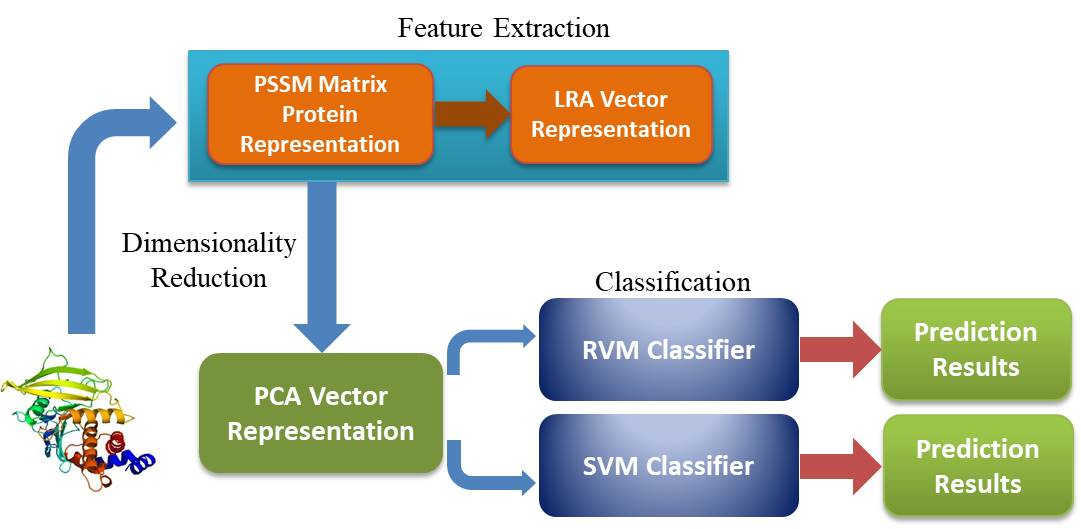
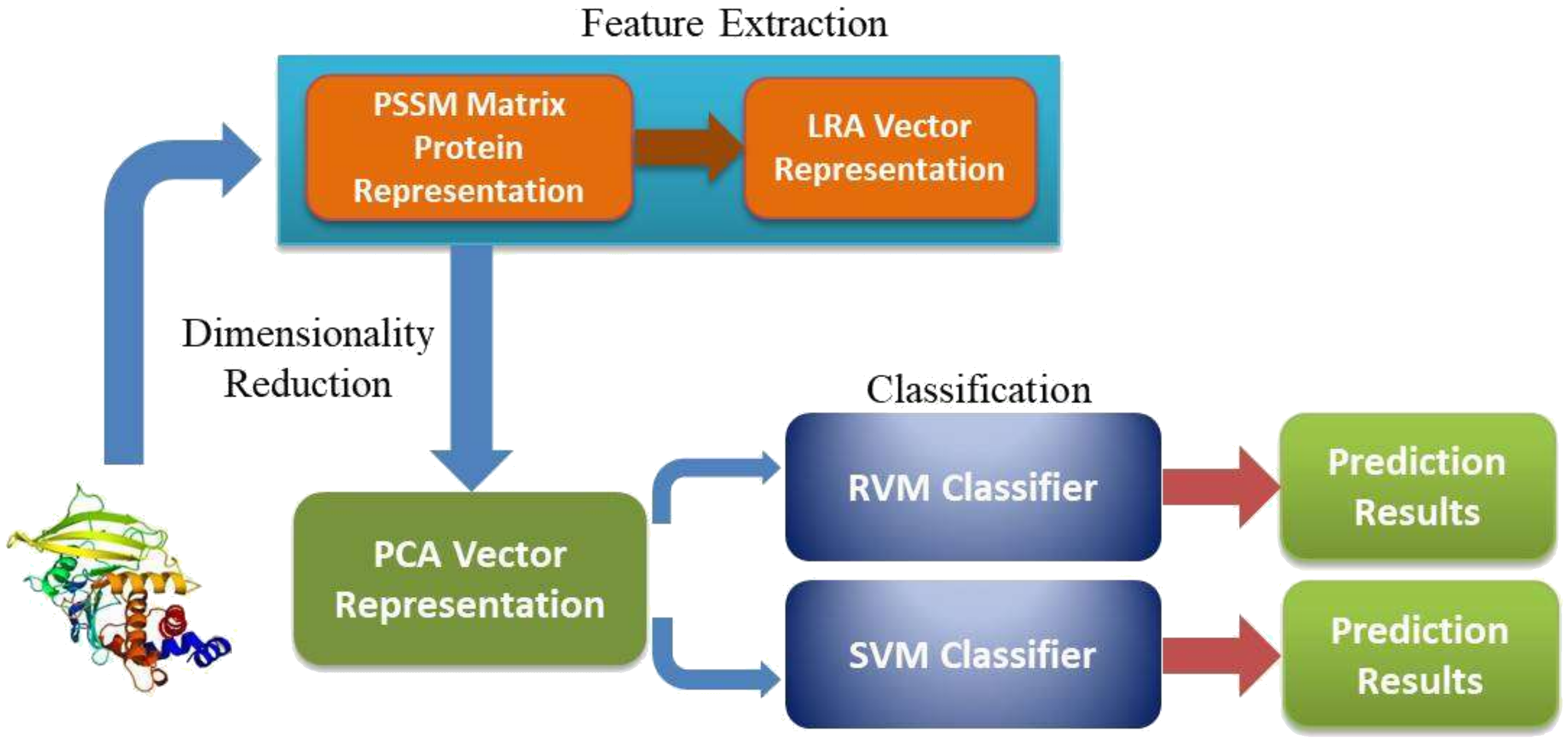
:
1. Introduction
2. Results and Discussion
2.1. Five-Fold Cross-Validation
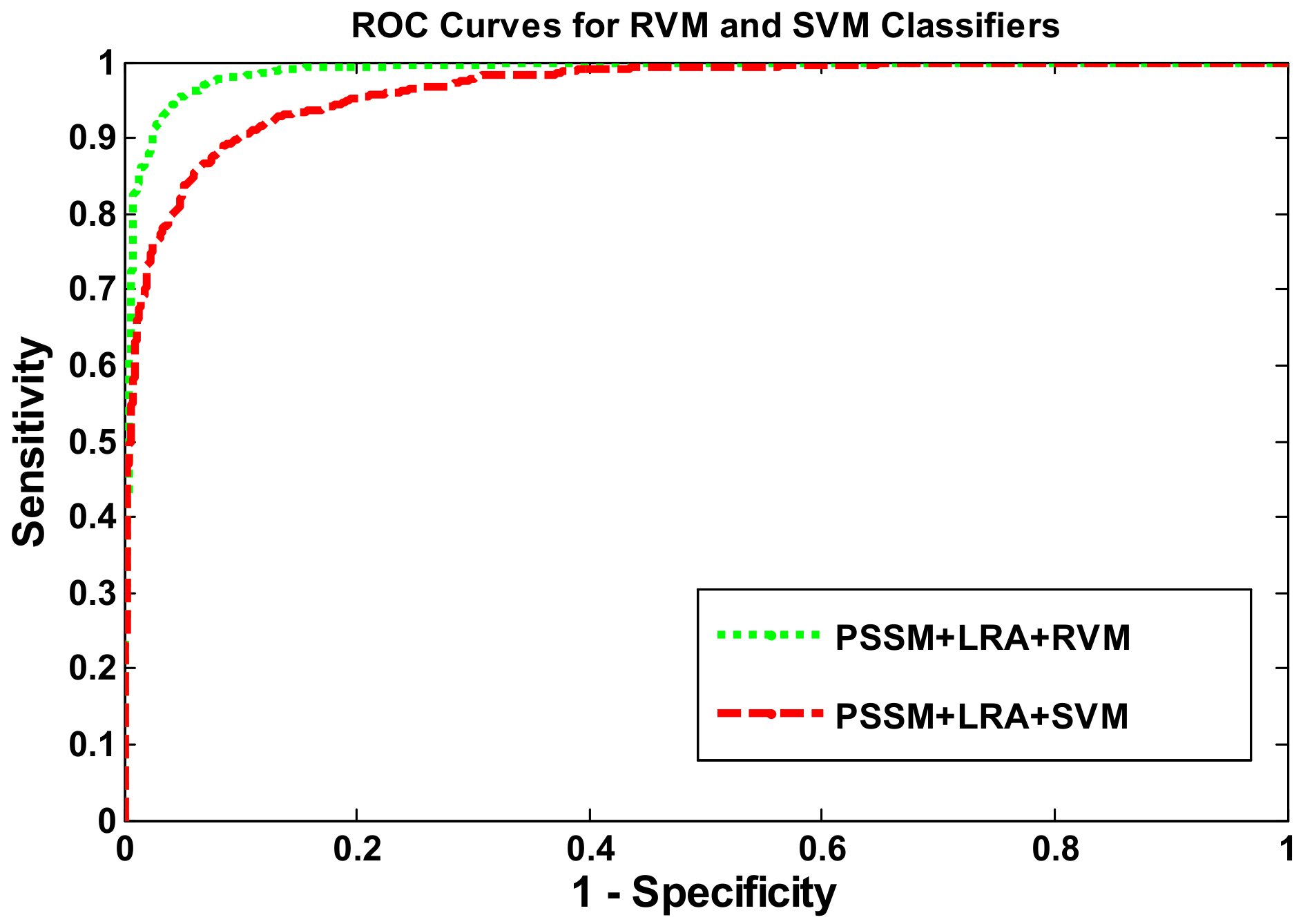
2.2. Comparison with the SVM-Based Approach Using the Same Feature Representation
2.3. A Comparison of the Proposed Method with Other Methods
2.4. An Assessment of the Prediction Performance on the Helicobacter pylori PPI Dataset
3. Materials and Methods
3.1. Dataset
3.2. Position Specific Scoring Matrix (PSSM)
3.3. Low-Rank Approximation (LRA)
3.4. Properties of the Proposed Algorithm
3.5. Relevance Vector Machine (RVM) Model
3.6. Procedure of the Proposed Method
3.7. Performance Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein-protein interactions in saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 7421, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.C.; Bosche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J.M.; Michon, A.M.; Cruciat, C.M.; et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Chiba, T.; Ozawa, R.; Yoshida, M.; Hattori, M.; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T. Global analysis of protein activities using proteome chips. Science 2001, 293, 2101–2105. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Puig, O.; Caspary, F.; Rigaut, G.; Rutz, B.; Bouveret, E.; Bragado-Nilsson, E.; Wilm, M.; Séraphin, B. The tandem affinity purification (tap) method: A general procedure of protein complex purification. Methods 2001, 24, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwínski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. Dip, the database of interacting proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. Mint: The molecular interaction database. Nucleic Acids Res. 2010, 40, D572–D574. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Donaldson, I.; Wolting, C.; Ouellette, B.F.F.; Pawson, T.; Hogue, C.W.V. Bind—The biomolecular interaction network database. Nucleic Acids Res. 2001, 29, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Helk, B.; Trout, B.L. A Computational Tool to Predict the Evolutionarily Conserved Protein-Protein Interaction Hot-Spot Residues from the Structure of the Unbound Protein; Asia Publishing Houser: Cambridge, MA, USA, 2014; pp. 326–333. [Google Scholar]
- Qiu, Z.; Wang, X. Prediction of protein-protein interaction sites using patch-based residue characterization. J. Theor. Biol. 2012, 293, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Zou, Q.; Xu, R.; Wang, X.; Chen, Q. Using distances between top-n-gram and residue pairs for protein remote homology detection. BMC Bioinform. 2014, 15, S3. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein remote homology detection by combining chou’s pseudo amino acid composition and profile-based protein representation. Mol. Inf. 2013, 32, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; You, Z.H.; Huang, D.S. Increasing the reliability of protein–protein interaction networks via non-convex semantic embedding. Neurocomputing 2013, 121, 99–107. [Google Scholar] [CrossRef]
- Huang, Q.; You, Z.; Zhang, X.; Zhou, Y. Prediction of protein–protein interactions with clustered amino acids and weighted sparse representation. Int. J. Mol. Sci. 2015, 16, 10855–10869. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-A.; You, Z.-H.; Gao, X.; Wong, L.; Wang, L. Using weighted sparse representation model combined with discrete cosine transformation to predict protein-protein interactions from protein sequence. BioMed Res. Int. 2015, 2015, 902198. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.-K.; You, Z.-H.; Ji, Z.; Zhu, L.; Huang, D.-S. Assessing and predicting protein interactions by combining manifold embedding with multiple information integration. BMC Bioinform. 2012, 13, S3. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ming, Z.; You, Z.; Li, S.; Xia, Y.; Leung, H. Improving network topology-based protein interactome mapping via collaborative filtering. Knowl. Based Syst. 2015, 90, 23–32. [Google Scholar] [CrossRef]
- You, Z.H.; Zhou, M.; Luo, X.; Li, S. Highly efficient framework for predicting interactions between proteins. IEEE Trans. Cybern. 2017, 47, 731–743. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Chan, K.C.; Hu, P. Predicting protein-protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest. PLoS ONE 2015, 10, e0125811. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Lei, Y.-K.; Gui, J.; Huang, D.-S.; Zhou, X. Using manifold embedding for assessing and predicting protein interactions from high-throughput experimental data. Bioinformatics 2010, 26, 2744–2751. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; You, Z.-H.; Huang, D.-S.; Wang, B. T-lse: A novel robust geometric approach for modeling protein-protein interaction networks. PLoS ONE 2013, 8, e58368. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Sarai, A. Pssm-based prediction of DNA binding sites in proteins. BMC Bioinform. 2005, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-W.; You, Z.-H.; Chen, X.; Li, L.-P.; Huang, D.-S.; Yan, G.-Y.; Nie, R.; Huang, Y.-A. Accurate prediction of protein-protein interactions by integrating potential evolutionary information embedded in pssm profile and discriminative vector machine classifier. Oncotarget 2017, 8, 23638. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. Hyperplanes for predicting protein–protein interactions. Neurocomputing 2005, 69, 257–263. [Google Scholar] [CrossRef]
- Nanni, L. Fusion of classifiers for predicting protein–protein interactions. Neurocomputing 2005, 68, 289–296. [Google Scholar] [CrossRef]
- Nanni, L. An ensemble of k-local hyperplanes for predicting protein-protein interactions. Neurocomputing 2006, 22, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-dpp: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, 384, 135–144. [Google Scholar] [CrossRef]
- Wang, Y.B.; You, Z.H.; Li, X.; Jiang, T.H.; Chen, X.; Zhou, X.; Wang, L. Predicting protein-protein interactions from protein sequences by a stacked sparse autoencoder deep neural network. Mol. Biosyst. 2017, 13, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Yang, Y.; Nishikawa, R.M.; Wernick, M.N.; Edwards, A. Relevance vector machine for automatic detection of clustered microcalcifications. IEEE Trans. Med. Imaging 2005, 24, 1278–1285. [Google Scholar] [PubMed]
- Widodo, A.; Kim, E.Y.; Son, J.D.; Yang, B.S.; Tan, A.C.C.; Gu, D.S.; Choi, B.K.; Mathew, J. Fault diagnosis of low speed bearing based on relevance vector machine and support vector machine. Expert Syst. Appl. 2009, 36, 7252–7261. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. Libsvm: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2001, 2, 1–27. [Google Scholar] [CrossRef]
- Yanzhi, G.; Lezheng, Y.; Zhining, W.; Menglong, L. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar]
- Zhou, Y.Z.; Gao, Y.; Zheng, Y.Y. Prediction of Protein-Protein Interactions Using Local Description of Amino Acid Sequence; Springer: Berlin/Heidelberg, Germany, 2011; pp. 254–262. [Google Scholar]
- Lei, Y.; Jun-Feng, X.; Jie, G. Prediction of protein-protein interactions from protein sequence using local descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar]
- You, Z.H.; Lei, Y.K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Roe, D.; Faulon, J.L. Predicting protein-protein interactions using signature products. Bioinformatics 2005, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Liberty, E.; Woolfe, F.; Martinsson, P.G.; Rokhlin, V.; Tygert, M. Randomized algorithms for the low-rank approximation of matrices. Proc. Natl. Acad. Sci. USA 2007, 104, 20167–20172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovsky, I. Structured low-rank approximation and its applications. Automatica 2008, 44, 891–909. [Google Scholar] [CrossRef]
- Tipping, M.E. Sparse bayesian learning and the relevance vector machine. J. Mach. Learn. Res. 2001, 1, 211–244. [Google Scholar]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. Halign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Zou, Q.; Ju, Y.; Li, D. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar]
- Zou, Q. Editorial (Thematic Issue: Machine Learning Techniques for Protein Structure, Genomics Function Analysis and Disease Prediction). Curr. Proteom. 2016, 13, 77–78. [Google Scholar] [CrossRef]
- Zou, Q.; Xie, S.; Lin, Z.; Wu, M.; Ju, Y. Finding the Best Classification Threshold in Imbalanced Classification. Big Data Res. 2016, 5, 2–8. [Google Scholar] [CrossRef]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, Z.; Xiao, L.; Xing, C.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; You, Z.H.; Li, L.P.; Huang, Y.A.; Yi, H.C. Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM. Molecules 2017, 22, 1366. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Model | Testing Set | Accuracy | Sensitivity | Specificity | PPV | NPV | MCC |
|---|---|---|---|---|---|---|---|
| PSSM+ LR+RVM | 1 | 94.7% | 95.4% | 94.0% | 93.9% | 95.46% | 89.3% |
| 2 | 95.3% | 96.1% | 94.5% | 94.7% | 95.96% | 91.1% | |
| 3 | 93.9% | 93.9% | 93.8% | 93.8% | 93.91% | 88.5% | |
| 4 | 93.8% | 93.6% | 94.1% | 94.4% | 93.22% | 88.4% | |
| 5 | 95.1% | 94.9% | 95.2% | 94.9% | 95.2% | 90.6% | |
| Average | 94.6 ± 0.6% | 94.8 ± 1.0% | 94.3 ± 0.5% | 94.3 ± 0.4% | 94.75 ± 1.1% | 89.6 ± 1.2% | |
| PSSM+ LR+SVM | 1 | 88.3% | 87.3% | 89.3% | 88.8% | 87.8% | 79.4% |
| 2 | 89.3% | 89.4% | 89.1% | 89.2% | 89.3% | 80.8% | |
| 3 | 89.8% | 89.2% | 90.3% | 90.7% | 88.8% | 81.6% | |
| 4 | 89.7% | 88.3% | 91.2% | 90.9% | 88.6% | 81.6% | |
| 5 | 90.0% | 88.4% | 91.5% | 90.8% | 89.2% | 81.9% | |
| Average | 89.4 ± 0.6% | 88.5 ± 0.8% | 90.3 ± 1.0% | 90.1 ± 1.0% | 88.7 ± 0.5% | 81.1 ± 1.0% |
| Model | Testing Set | Acc (%) | Sen (%) | Pre (%) | Mcc (%) |
|---|---|---|---|---|---|
| Guos’ work [35] | ACC | 89.3 ± 2.6 | 89.9 ± 3.6 | 88.8 ± 6.1 | N/A |
| AC | 87.4 ± 1.3 | 87.3 ± 4.6 | 87.8 ± 4.3 | N/A | |
| Zhous’ work [36] | SVM+LD | 88.6 ± 0.3 | 87.4 ± 0.2 | 89.5 ± 0.6 | 77.2 ± 0.7 |
| Yangs’ work [37] | Cod1 | 75.1 ± 1.1 | 75.8 ± 1.2 | 74.8 ± 1.2 | N/A |
| Cod2 | 80.0 ± 1.0 | 76.8 ± 0.6 | 82.2 ± 1.3 | N/A | |
| Cod3 | 80.4 ± 0.4 | 78.1 ± 0.9 | 81.7 ± 0.9 | N/A | |
| Cod4 | 86.2 ± 1.1 | 81.0 ± 1.7 | 90.2 ± 1.3 | N/A | |
| Yous’ work [38] | PCA-EELM | 87.0 ± 0.2 | 86.2 ± 0.4 | 87.6 ± 0.3 | 77.4 ± 0.4 |
| Proposed method | LRA+RVM | 94.6 ± 0.6 | 94.8 ± 1.0 | 94.4 ± 0.4 | 89.6 ± 1.2 |
| Methods | Acc(%) | Sen (%) | Pre (%) | Mcc (%) |
|---|---|---|---|---|
| HKNN | 84.0 | 86.0 | 84.0 | N/A |
| Phylogenetic bootstrap | 75.8 | 69.8 | 80.2 | N/A |
| Signature Products | 83.4 | 79.9 | 85.7 | N/A |
| Boosting | 79.5 | 80.4 | 81.7 | N/A |
| Proposed method | 84.7 ± 1.0 | 84.4 ± 1.2 | 85.9 ± 0.8 | 76.7 ± 1.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.-P.; Wang, Y.-B.; You, Z.-H.; Li, Y.; An, J.-Y. PCLPred: A Bioinformatics Method for Predicting Protein–Protein Interactions by Combining Relevance Vector Machine Model with Low-Rank Matrix Approximation. Int. J. Mol. Sci. 2018, 19, 1029. https://doi.org/10.3390/ijms19041029
Li L-P, Wang Y-B, You Z-H, Li Y, An J-Y. PCLPred: A Bioinformatics Method for Predicting Protein–Protein Interactions by Combining Relevance Vector Machine Model with Low-Rank Matrix Approximation. International Journal of Molecular Sciences. 2018; 19(4):1029. https://doi.org/10.3390/ijms19041029
Chicago/Turabian StyleLi, Li-Ping, Yan-Bin Wang, Zhu-Hong You, Yang Li, and Ji-Yong An. 2018. "PCLPred: A Bioinformatics Method for Predicting Protein–Protein Interactions by Combining Relevance Vector Machine Model with Low-Rank Matrix Approximation" International Journal of Molecular Sciences 19, no. 4: 1029. https://doi.org/10.3390/ijms19041029