Tau-Induced Pathology in Epilepsy and Dementia: Notions from Patients and Animal Models

 ,
,
Abstract
:1. Epilepsy in Dementia
2. AD and Seizures
3. Seizures in FTDP-17 and Other Dementias
3.1. FTDP-17 and Seizures
3.2. Seizures in PSP
3.3. Seizures in DLB
3.4. Epilepsy in DS
4. Animal Models
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Forsgren, L.; Edvinsson, S.O.; Blomquist, H.K.; Heijbel, J.; Sidenvall, R. Epilepsy in a population of mentally retarded children and adults. Epilepsy Res. 1990, 6, 234–248. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Hauser, W.A.; Annegers, J.F.; Kokmen, E.; Rocca, W.A. Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Sherzai, D.; Losey, T.; Vega, S.; Sherzai, A. Seizures and dementia in the elderly: Nationwide Inpatient Sample 1999–2008. Epilepsy Behav. 2014, 36, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F.; Catanzaro, P.; Doss, R.C.; ARguello, R.; Frey, W.H. Seizures in Alzheimer’s disease: Clinicopathologic study. J. Geriatr. Psychiatry Neurol. 1994, 7, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Honig, L.S.; Choi, H.; Cantero, J.; Brandt, J.; Blacker, D.; Albert, M.; Amatniek, J.C.; Marder, K.; Bell, K.; et al. Seizures in Alzheimer disease: Who, when, and how common? Arch. Neurol. 2009, 66, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.F.; Morris, J.C.; Ashkin, K.; Coben, L.A. Advanced Alzheimer’s disease is a risk factor for late-onset seizures. Arch. Neurol. 1990, 47, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, M. Down syndrome, Alzheimer’s disease and seizures. Brain Dev. 2005, 27, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Jin, S.; He, F.; Emond, J.A.; Raman, R.; Thomas, R.G.; Sano, M.; Quinn, J.F.; Tariot, P.N.; Galasko, D.R.; et al. Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, N.; Assal, F.; Seeck, M. From here to epilepsy: The risk of seizure in patients with Alzheimer’s disease. Epileptic Disord. 2016, 18, 1–12. [Google Scholar] [PubMed]
- Beagle, A.J.; Darwish, S.M.; Ranasinghe, K.G.; La, A.L.; Karageorgiou, E.; Vossel, K.A. Relative Incidence of Seizures and Myoclonus in Alzheimer’s Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J. Alzheimers Dis. 2017, 60, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, T.G.; Duvoisin, R.C.; Manocha, M.; Chokroverty, S. Seizures in progressive supranuclear palsy. Neurology 1989, 39, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.A.; Morris, M.L.; Heston, L.L.; Anderson, V.E. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986, 36, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Stern, Y.; Sano, M.; Mayeux, R. Cumulative risks of developing extrapyramidal signs, psychosis, or myoclonus in the course of Alzheimer’s disease. Arch. Neurol. 1991, 48, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Alladi, S.; Xuereb, J.; Bak, T.; Nestor, P.; Knibb, J.; Patterson, K.; Hodges, J.R. Focal cortical presentations of Alzheimer’s disease. Brain 2007, 130, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Rabinovici, G.D.; Mayo, M.C.; Wilson, S.M.; Seeley, W.W.; DeArmond, S.J.; Huang, E.J.; Trojanowski, J.Q.; Growdon, M.E.; Jang, J.Y.; et al. Clinicopathological correlations in corticobasal degeneration. Ann. Neurol. 2011, 70, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszek, J.; Lang, A.E.; Jankovic, J.; Greene, P.; Deck, J. What is it? Case 1, 1994: Rapidly progressive aphasia, apraxia, dementia, myoclonus, and parkinsonism. Mov. Disord. 1994, 9, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, O. FTDP-17: Phenotypical heterogeneity within P301S. Ann. Neurol. 2000, 48, 126. [Google Scholar] [CrossRef]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Tacik, P.; Sanchez-Contreras, M.; DeTure, M.; Murray, M.E.; Rademakers, R.; Ross, O.A.; Wszolek, Z.K.; Parisi, J.E.; Knopman, D.S.; Petersen, R.C.; et al. Clinicopathologic heterogeneity in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) due to microtubule-associated protein tau (MAPT) p.P301L mutation, including a patient with globular glial tauopathy. Neuropathol. Appl. Neurobiol. 2017, 43, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Larner, A.J. Epileptic seizures in AD patients. Neuromol. Med. 2010, 12, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Lozsadi, D.A.; Larner, A.J. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Noebels, J. A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia 2011, 52 (Suppl. 1), 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra, M.; Carnero, C.; Blesa, R.; Gelpí, J.L.; Ballesta, F.; Oliva, R. A presenilin 1 mutation (Ser169Pro) associated with early-onset AD and myoclonic seizures. Neurology 1999, 52, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Brice, A.; Hannequin, D.; Tardieu, S.; Dubois, B.; Calenda, A.; Brun, E.; Penet, C.; Tayot, J.; Martinez, M. A large pedigree with early-onset Alzheimer’s disease: Clinical, neuropathologic, and genetic characterization. Neurology 1995, 45, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Filla, A.; De Michele, G.; Cocozza, S.; Patrignani, A.; Volpe, G.; Castaldo, I.; Ruggiero, G.; Bonavita, V.; Masters, C.; Casari, G.; et al. Early onset autosomal dominant dementia with ataxia, extrapyramidal features, and epilepsy. Neurology 2002, 58, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.G.; Nielsen, J.E.; Stokholm, J.; Schwartz, M.; Batbayli, M.; Ballegaard, M.; Erdal, J.; Krabbe, K.; Waldemar, G. Atypical early-onset Alzheimer’s disease caused by the Iranian APP mutation. J. Neurol. Sci. 2008, 268, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Larner, A.J.; Doran, M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J. Neurol. 2006, 253, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Cabrejo, L.; Guyant-Maréchal, L.; Laquerrière, A.; Vercelletto, M.; De la Fournière, F.; Thomas-Antérion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.J.; Norton, J.; Coats, M.A.; Chakraverty, S.; Hou, C.E.; Jervis, R.; Lendon, C.L.; Goate, A.M.; McKeel, D.W.; Morris, J.C. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch. Neurol. 2005, 62, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Van Swieten, J.C.; Stevens, M.; Rosso, S.M.; Rizzu, P.; Joosse, M.; de Koning, I.; Kamphorst, W.; Ravid, R.; Spillantini, M.G.; Niermeijer, M.F.; et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann. Neurol. 1999, 46, 617–626. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Le Guennec, K.; Quenez, O.; Nicolas, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; Alexander, J.; Paschou, P.; Charbonnier, C.; Bellenguez, C.; et al. 17q21.31 duplication causes prominent tau-related dementia with increased MAPT expression. Mol. Psychiatry 2017, 22, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid beta-protein and the genetics of Alzheimer’s disease. J. Biol. Chem. 1996, 271, 18295–18298. [Google Scholar] [CrossRef] [PubMed]
- Zarea, A.; Charbonnier, C.; Rovelet-Lecrux, A.; Nicolas, G.; Rousseau, S.; Borden, A.; Pariente, J.; Le Ber, I.; Pasquier, F.; Formaglio, M.; et al. Seizures in dominantly inherited Alzheimer disease. Neurology 2016, 87, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.F.; Chu, L.W.; Chan, A.O.; Ha, J.; Li, Y.; Song, Y.Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: A case series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arrastia, R.; Gong, Y.; Fair, S.; Scott, K.D.; Garcia, M.C.; Carlile, M.C.; Agostini, M.A.; Van Ness, P.C. Increased risk of late posttraumatic seizures associated with inheritance of APOE epsilon4 allele. Arch. Neurol. 2003, 60, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Erkinjuntti, T.; Ostbye, T.; Steenhuis, R.; Hachinski, V. The effect of different diagnostic criteria on the prevalence of dementia. N. Engl. J. Med. 1997, 337, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Clemens, Z.; Janszky, J.; Szucs, A.; Békésy, M.; Clemens, B.; Halász, P. Interictal epileptic spiking during sleep and wakefulness in mesial temporal lobe epilepsy: A comparative study of scalp and foramen ovale electrodes. Epilepsia 2003, 44, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, D.; Fohlen, M.; Jalin, C.; Dorfmuller, G.; Bulteau, C.; Delalande, O. Foramen ovale electrodes in the preoperative evaluation of temporal lobe epilepsy in children. Epilepsia 2009, 50, 2085–2096. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Honig, L.S.; Scarmeas, N. Seizures and epilepsy in Alzheimer’s disease. CNS Neurosci. Ther. 2012, 18, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Subota, A.; Pham, T.; Jetté, N.; Sauro, K.; Lorenzetti, D.; Holroyd-Leduc, J. The association between dementia and epilepsy: A systematic review and meta-analysis. Epilepsia 2017, 58, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Montejo de Garcini, E.; Serrano, L.; Avila, J. Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem. Biophys. Res. Commun. 1986, 141, 790–796. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Iqbal, K.; Wiśniewski, H.M.; Shelanski, M.L.; Brostoff, S.; Liwnicz, B.H.; Terry, R.D. Protein changes in senile dementia. Brain Res. 1974, 77, 337–343. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I.; Zaidi, T.; Merz, P.A.; Wen, G.Y.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtubule assembly in Alzheimer’s disease. Lancet 1986, 2, 421–426. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B.A. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Horváth, A.; Szűcs, A.; Barcs, G.; Noebels, J.L.; Kamondi, A. Epileptic Seizures in Alzheimer Disease: A Review. Alzheimer Dis. Assoc. Disord. 2016, 30, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ou, S.; Yin, M.; Xu, T.; Wang, T.; Liu, Y.; Ding, X.; Yu, X.; Yuan, J.; Huang, H.; et al. N-methyl-d-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3beta and cyclin-dependent kinase 5. Discov. Med. 2017, 23, 221–234. [Google Scholar] [PubMed]
- Szot, P. Common factors among Alzheimer’s disease, Parkinson’s disease, and epilepsy: Possible role of the noradrenergic nervous system. Epilepsia 2012, 53 (Suppl. 1), 61–66. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. Alzheimer’s disease and epilepsy: Insight from animal models. Future Neurol. 2012, 7, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Müller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Devidze, N.; Verret, L.; Ho, K.; Halabisky, B.; Thwin, M.T.; Kim, D.; Hamto, P.; Lo, I.; Yu, G.Q.; et al. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 2010, 68, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Thom, M.; Martinian, L.; Harding, B.; Cross, J.H.; Nikolic, M.; Sisodiya, S.M. Pathological tau tangles localize to focal cortical dysplasia in older patients. Epilepsia 2007, 48, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Smith, A.C.; Humphreys, P.; Schwartzentruber, J.; Beaulieu, C.L.; Bulman, D.E.; Majewski, J.; Woulfe, J.; Michaud, J.; Boycott, K.M.; et al. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol. Aging 2015, 36, 1222.e1–1222.e5. [Google Scholar] [CrossRef] [PubMed]
- Tai, X.Y.; Koepp, M.; Duncan, J.S.; Fox, N.; Thompson, P.; Baxendale, S.; Liu, J.Y.; Reeves, C.; Michalak, Z.; Thom, M. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: A study of temporal lobe resections. Brain 2016, 139, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Russin, J.; Heck, C.; Kawata, K.; Adiga, R.; Yen, W.; Lambert, J.; Stear, B.; Law, M.; Marquez, Y.; et al. Dysregulation of PINCH signaling in mesial temporal epilepsy. J. Clin. Neurosci. 2017, 36, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Thom, M.; Liu, J.Y.; Thompson, P.; Phadke, R.; Narkiewicz, M.; Martinian, L.; Marsdon, D.; Koepp, M.; Caboclo, L.; Catarino, C.B.; et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: A post-mortem study. Brain 2011, 134, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Biel, N.; Canudas, A.M.; Camins, A.; Pallàs, M. Kainate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem. Int. 2007, 50, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J. Alzheimers Dis. 2009, 17, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.F.; Zeng, C.; Ma, Y.F.; Guo, T.H.; Chen, J.M.; Chen, Y.; Cai, X.F.; Li, F.R.; Wang, X.H.; Huang, W.J.; et al. Potential roles of Cdk5/p35 and tau protein in hippocampal mossy fiber sprouting in the PTZ kindling model. Clin. Lab. 2010, 56, 127–136. [Google Scholar] [PubMed]
- Liu, J.; Wang, L.N.; Wu, L.Y.; Wang, Y.P. Treatment of epilepsy for people with Alzheimer’s disease. Cochrane Database Syst. Rev. 2016, 11, CD011922. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M. Disruption of the cytoskeleton in Alzheimer’s disease. Curr. Opin. Neurobiol. 1995, 5, 663–668. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ishiguro, K.; Uchida, T.; Takashima, A.; Lemere, C.A.; Imahori, K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 1996, 92, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Shiomura, Y.; Okabe, S. Tau proteins: The molecular structure and mode of binding on microtubules. J. Cell Biol. 1988, 107, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 1998, 93, 1147–1158. [Google Scholar] [CrossRef]
- Collot, M.; Louvard, D.; Singer, S.J. Lysosomes are associated with microtubules and not with intermediate filaments in cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1984, 81, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, P.; Detrez, J.R.; Verschuuren, M.; Kuijlaars, J.; Nuydens, R.; Timmermans, J.P.; De Vos, W.H. Dysregulation of Microtubule Stability Impairs Morphofunctional Connectivity in Primary Neuronal Networks. Front. Cell. Neurosci. 2017, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 1978, 2, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Katzman, R.; Terry, R.D. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature 1980, 288, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Blessed, G.; Tomlinson, B.E.; Perry, R.H.; Crow, T.J.; Cross, A.J.; Dockray, G.J.; Dimaline, R.; Arregui, A. Neurochemical activities in human temporal lobe related to aging and Alzheimer-type changes. Neurobiol. Aging 1981, 2, 251–256. [Google Scholar] [CrossRef]
- Strac, D.S.; Muck-Seler, D.; Pivac, N. Neurotransmitter measures in the cerebrospinal fluid of patients with Alzheimer’s disease: A review. Psychiatr. Danub. 2015, 27, 14–24. [Google Scholar] [PubMed]
- García-Ayllón, M.S.; Small, D.H.; Avila, J.; Sáez-Valero, J. Revisiting the Role of Acetylcholinesterase in Alzheimer’s Disease: Cross-Talk with P-tau and β-Amyloid. Front. Mol. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Baskys, A.; Reynolds, J.N.; Carlen, P.L. NMDA depolarizations and long-term potentiation are reduced in the aged rat neocortex. Brain Res. 1990, 530, 142–146. [Google Scholar] [CrossRef]
- Tang, B.L. Neuronal protein trafficking associated with Alzheimer disease: From APP and BACE1 to glutamate receptors. Cell Adh. Migr. 2009, 3, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, W.; Yan, Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, E.; Glazier, S.; Bell, W.; Tytell, M.; Nosel, E.; Pons, T.; Deadwyler, S.A. Changes in glutamate receptor subunit composition in hippocampus and cortex in patients with refractory epilepsy. J. Neurol. Sci. 1997, 153, 35–45. [Google Scholar] [CrossRef]
- Pooler, A.M.; Noble, W.; Hanger, D.P. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 2014, 76 Pt A, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [PubMed]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.J.; Davis, M.J.; Zawieja, D.C.; Muthuchamy, M. Calcium sensitivity and cooperativity of permeabilized rat mesenteric lymphatics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1524–R1532. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.L.; Ferreira, A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-d-aspartate receptors in hippocampal neurons. J. Biol. Chem. 2006, 281, 28079–28089. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.; Mezler, M.; Müller, M.K.; Wicke, K.; Gross, G.; Draguhn, A.; Bruehl, C.; Nimmrich, V. Synthetic Aβ oligomers (Aβ(1-42) globulomer) modulate presynaptic calcium currents: Prevention of Aβ-induced synaptic deficits by calcium channel blockers. Eur. J. Pharmacol. 2013, 702, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, M.; Henderson, Z.; Pearson, H.A. Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1-40) is dependent on solubility status. Brain Res. 2002, 956, 254–261. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.P.; Shapiro, M.L. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Nimmrich, V.; Ebert, U. Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev. Neurosci. 2009, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cretin, B.; Philippi, N.; Dibitonto, L.; Blanc, F. Epilepsy at the prodromal stages of neurodegenerative diseases. Geriatr. Psychol. Neuropsychiatr. Vieil. 2017, 15, 75–82. [Google Scholar] [PubMed]
- Leonard, A.S.; McNamara, J.O. Does epileptiform activity contribute to cognitive impairment in Alzheimer’s disease? Neuron 2007, 55, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Damar, U.; Gersner, R.; Johnstone, J.T.; Schachter, S.; Rotenberg, A. Huperzine A: A promising anticonvulsant, disease modifying, and memory enhancing treatment option in Alzheimer’s disease. Med. Hypotheses 2017, 99, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, E.; Ligori, L.D. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010, 17, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Werhahn, K.J.; Trinka, E.; Dobesberger, J.; Unterberger, I.; Baum, P.; Deckert-Schmitz, M.; Kniess, T.; Schmitz, B.; Bernedo, V.; Ruckes, C.; et al. A randomized, double-blind comparison of antiepileptic drug treatment in the elderly with new-onset focal epilepsy. Epilepsia 2015, 56, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed]
- Armon, C.; Peterson, G.W.; Liwnicz, B.H. Alzheimer’s disease underlies some cases of complex partial status epilepticus. J. Clin. Neurophysiol. 2000, 17, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.C.; Dove, G.; Cascino, G.D.; Petersen, R.C. Recurrent seizures in patients with dementia: Frequency, seizure types, and treatment outcome. Epilepsy Behav. 2009, 14, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Gais, S.; Born, J. Declarative memory consolidation: Mechanisms acting during human sleep. Learn. Mem. 2004, 11, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Whatley, V.J.; Harris, R.A. The cytoskeleton and neurotransmitter receptors. Int. Rev. Neurobiol. 1996, 39, 113–143. [Google Scholar] [PubMed]
- Gleichmann, M.; Mattson, M.P. Alzheimer’s disease and neuronal network activity. Neuromol. Med. 2010, 12, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Mattson, M.P. Adverse stress, hippocampal networks, and Alzheimer’s disease. Neuromol. Med. 2010, 12, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Joshi, R.; Medhi, B. Cracking novel shared targets between epilepsy and Alzheimer’s disease: Need of the hour. Rev. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Morris, J.H.; Kosik, K.S.; Selkoe, D.J. Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol. 1987, 22, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hansen, L.A.; Vincent, I.; Mallory, M.; Masliah, E. Neurofibrillary tangles in the dentate granule cells of patients with Alzheimer’s disease, Lewy body disease and progressive supranuclear palsy. Acta Neuropathol. 1997, 93, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Ruan, D.; Crystal, H.; Mark, M.H.; Davies, P.; Kress, Y.; Yen, S.H. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: Light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology 1991, 41, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D. Neuronal fibrous protein in human pathology. J. Neuropathol. Exp. Neurol. 1971, 30, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Wilhelmsen, K.; Sima, A.A.; Jones, M.Z.; D’Amato, C.J.; Gilman, S. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference Participants. Ann. Neurol. 1997, 41, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Wszolek, Z.K.; Tsuboi, Y.; Ghetti, B.; Pickering-Brown, S.; Baba, Y.; Cheshire, W.P. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis. 2006, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006254. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Bird, T.D.; Ghetti, B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: A new group of tauopathies. Brain Pathol. 1998, 8, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Takamatsu, J.; D’Souza, I.; Crowther, R.A.; Kawamata, T.; Hasegawa, M.; Hasegawa, H.; Spillantini, M.G.; Tanimukai, S.; Poorkaj, P.; et al. A novel mutation at position +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto). Ann. Neurol. 2000, 47, 422–429. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Van Swieten, J.C.; Goedert, M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2000, 2, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Lim, F.; Arrasate, M.; Avila, J. The FTDP-17-linked mutation R406W abolishes the interaction of phosphorylated tau with microtubules. J. Neurochem. 2000, 74, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Götz, J.; Wiederhold, K.H.; Tolnay, M.; Mistl, C.; Jaton, A.L.; Hong, M.; Ishihara, T.; Lee, V.M.; Trojanowski, J.Q.; et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Hernández, F.; Lucas, J.J.; Gómez-Ramos, P.; Morán, M.A.; Avila, J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Engel, T.; Lucas, J.J.; Gómez-Ramos, P.; Moran, M.A.; Avila, J.; Hernández, F. Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol. Aging 2006, 27, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martin, M.; Hernandez, F.; Avila, J. Expression of frontotemporal dementia with parkinsonism associated to chromosome 17 tau induces specific degeneration of the ventral dentate gyrus and depressive-like behavior in mice. Neuroscience 2011, 196, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cabrero, A.M.; Guerrero-Lopez, R.; Giraldez, B.G.; Llorens-Martin, M.; Avila, J.; Serratosa, J.M.; Sanchez, M.P. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 2013, 58, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.C.; Richardson, J.C.; Olszewski, J. Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch. Neurol. 1964, 10, 333–359. [Google Scholar] [CrossRef] [PubMed]
- Litvan, I.; Agid, Y.; Calne, D.; Campbell, G.; Dubois, B.; Duvoisin, R.C.; Goetz, C.G.; Golbe, L.I.; Grafman, J.; Growdon, J.H.; et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): Report of the NINDS-SPSP international workshop. Neurology 1996, 47, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Valpuesta, J.M.; de Garcini, E.M.; Quintana, C.; Arrasate, M.; López Carrascosa, J.L.; Rábano, A.; García de Yébenes, J.; Avila, J. Ferritin is associated with the aberrant tau filaments present in progressive supranuclear palsy. Am. J. Pathol. 1998, 152, 1531–1539. [Google Scholar] [PubMed]
- Dickson, D.W.; Ahmed, Z.; Algom, A.A.; Tsuboi, Y.; Josephs, K.A. Neuropathology of variants of progressive supranuclear palsy. Curr. Opin. Neurol. 2010, 23, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Langui, D.; Lautenschlager, C.; Ulrich, J.; Brion, J.P.; Anderton, B.H. Progressive supranuclear palsy: Extensive neuropil threads in addition to neurofibrillary tangles. Very similar antigenicity of subcortical neuronal pathology in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. 1988, 77, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.P.; Gonzalo, I.; Avila, J.; De Yebenes, J.G. Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J. Alzheimers Dis. 2002, 4, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Morales, B.; Martinez, A.; Gonzalo, I.; Vidal, L.; Ros, R.; Gomez-Tortosa, E.; Rabano, A.; Ampuero, I.; Sanchez, M.; Hoenicka, J.; et al. Steele-Richardson-Olszewski syndrome in a patient with a single C212Y mutation in the parkin protein. Mov. Disord. 2002, 17, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Lantos, P.; Stratton, M.; Roques, P.; Rossor, M. Familial progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 1993, 56, 473–476. [Google Scholar] [CrossRef] [PubMed]
- De Yébenes, J.G.; Sarasa, J.L.; Daniel, S.E.; Lees, A.J. Familial progressive supranuclear palsy. Description of a pedigree and review of the literature. Brain 1995, 118 Pt 5, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.; Pernaute, R.S.; Fontán, A.; Ruíz, P.G.; Honnorat, J.; Lynch, T.; Chin, S.; Gonzalo, I.; Rábano, A.; Martínez, A.; et al. Clinical genetics of familial progressive supranuclear palsy. Brain 1999, 122 Pt 7, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Melhem, N.M.; Dickson, D.W.; Sleiman, P.M.; Wang, L.S.; Klei, L.; Rademakers, R.; de Silva, R.; Litvan, I.; Riley, D.E.; et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011, 43, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Lees, A.J. Progressive supranuclear palsy: Clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009, 8, 270–279. [Google Scholar] [CrossRef]
- Fujioka, S.; Van Gerpen, J.A.; Uitti, R.J.; Dickson, D.W.; Wszolek, Z.K. Familial progressive supranuclear palsy: A literature review. Neurodegener. Dis. 2014, 13, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Tolosa, E.; Ezquerra, M.; Molinuevo, J.L.; Valldeoriola, F.; Burguera, J.; Calopa, M.; Villa, M.; Ballesta, F. Significant changes in the tau A0 and A3 alleles in progressive supranuclear palsy and improved genotyping by silver detection. Arch. Neurol. 1998, 55, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Stanford, P.M.; Halliday, G.M.; Brooks, W.S.; Kwok, J.B.; Storey, C.E.; Creasey, H.; Morris, J.G.; Fulham, M.J.; Schofield, P.R. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: Expansion of the disease phenotype caused by tau gene mutations. Brain 2000, 123 Pt 5, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Donker Kaat, L.; Boon, A.J.; Azmani, A.; Kamphorst, W.; Breteler, M.M.; Anar, B.; Heutink, P.; van Swieten, J.C. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 2009, 73, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Pastor, P.; Pastor, E.; Carnero, C.; Vela, R.; García, T.; Amer, G.; Tolosa, E.; Oliva, R. Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann. Neurol. 2001, 49, 263–267. [Google Scholar] [CrossRef]
- Ros, R.; Thobois, S.; Streichenberger, N.; Kopp, N.; Sanchez, M.P.; Perez, M.; Hoenicha, J.; Avila, J.; Honnorat, J.; de Yebenes, J.G. A new mutation of the tau gene, G303V, in early-onset familial progressive supranuclear palsy. Arch. Neurol. 2005, 62, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Im, S.Y.; Kim, Y.E.; Kim, Y.J. Genetics of Progressive Supranuclear Palsy. J. Mov. Disord. 2015, 8, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, T.; Landau, W.M.; Torack, R.M. Progressive supranuclear palsy with action myoclonus, seizures. Neurology 1974, 24, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Duvoisin, R.C.; Golbe, L.I.; Lepore, F.E. Progressive supranuclear palsy. Can. J. Neurol. Sci. 1987, 14, 547–554. [Google Scholar] [PubMed]
- Lanza, G.; Papotto, M.; Pennisi, G.; Bella, R.; Ferri, R. Epileptic seizure as a precipitating factor of vascular progressive supranuclear palsy: A case report. J. Stroke Cerebrovasc. Dis. 2014, 23, e379–e381. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Stephens, S.; Ballard, C. Dementia with Lewy bodies: Clinical features and treatment. Drugs Aging 2001, 18, 397–407. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 1996, 47, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Clarimón, J.; Molina-Porcel, L.; Gómez-Isla, T.; Blesa, R.; Guardia-Laguarta, C.; González-Neira, A.; Estorch, M.; Ma Grau, J.; Barraquer, L.; Roig, C.; et al. Early-onset familial lewy body dementia with extensive tauopathy: A clinical, genetic, and neuropathological study. J. Neuropathol. Exp. Neurol. 2009, 68, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Yoo, S.W.; Lee, K.S.; Kim, J.S. Epileptic seizure presenting as dementia with Lewy bodies. Gen. Hosp. Psychiatry 2014, 36, 230.e3–230.e5. [Google Scholar] [CrossRef] [PubMed]
- Ukai, K.; Fujishiro, H.; Watanabe, M.; Kosaka, K.; Ozaki, N. Similarity of symptoms between transient epileptic amnesia and Lewy body disease. Psychogeriatrics 2017, 17, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Gholipour, T.; Mitchell, S.; Sarkis, R.A.; Chemali, Z. The clinical and neurobehavioral course of Down syndrome and dementia with or without new-onset epilepsy. Epilepsy Behav. 2017, 68, 11–16. [Google Scholar] [CrossRef] [PubMed]
- D’ORSI, G.; Specchio, L.M.; Epilepsy, A.S.G.o.S.M. Progressive myoclonus epilepsy in Down syndrome patients with dementia. J. Neurol. 2014, 261, 1584–1597. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Doran, E.; Nguyen, V.Q.; Tournay, A.; Movsesyan, N.; Gillen, D.L. Down syndrome and dementia: Seizures and cognitive decline. J. Alzheimers Dis. 2012, 29, 177–185. [Google Scholar] [PubMed]
- De Simone, R.; Puig, X.S.; Gélisse, P.; Crespel, A.; Genton, P. Senile myoclonic epilepsy: Delineation of a common condition associated with Alzheimer’s disease in Down syndrome. Seizure 2010, 19, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Evenhuis, H.M. The natural history of dementia in Down’s syndrome. Arch. Neurol. 1990, 47, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Williams, R.S. A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 1989, 46, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Zis, P.; Strydom, A.; Buckley, D.; Adekitan, D.; McHugh, P.C. Cognitive ability in Down syndrome and its relationship to urinary neopterin, a marker of activated cellular immunity. Neurosci. Lett. 2017, 636, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K.; Ho, K.W.; Singh, I. Age of seizure onset in adults with Down’s syndrome. Int. J. Clin. Pract. 2001, 55, 442–444. [Google Scholar] [PubMed]
- Zis, P.; Strydom, A. Clinical aspects and biomarkers of Alzheimer’s disease in Down syndrome. Free Radic. Biol. Med. 2018, 114, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Paitel, E.; Kawarabayashi, T.; Ikeda, M.; Chishti, M.A.; Janus, C.; Matsubara, E.; Sasaki, A.; Kawarai, T.; Phinney, A.L.; et al. Cortical neuronal and glial pathology in TgTauP301L transgenic mice: Neuronal degeneration, memory disturbance, and phenotypic variation. Am. J. Pathol. 2006, 169, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Navarro, P.; Gallego, E.; Avila, J.; de Yebenes, J.G.; Sanchez, M.P. Park2-null/tau transgenic mice reveal a functional relationship between parkin and tau. J. Alzheimers Dis. 2008, 13, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Navarro, P.; Gallego, E.; Garcia-Cabrero, A.M.; Avila, J.; Sanchez, M.P. Hyperphosphorylated tau aggregates in the cortex and hippocampus of transgenic mice with mutant human FTDP-17 Tau and lacking the PARK2 gene. Acta Neuropathol. 2009, 117, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H.; He, P.; Lee, V.M.; Woolf, C.J.; et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Tartaglia, M.C.; Yener, G.; Genç, S.; Seeley, W.W.; Sanchez-Juan, P.; Moreno, F.; Mendez, M.F.; Klein, E.; Rademakers, R.; et al. Neurodegenerative disease phenotypes in carriers of MAPT p.A152T, a risk factor for frontotemporal dementia spectrum disorders and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2013, 27, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Wöhrer, A.; Ströbel, T.; Botond, G.; Attems, J.; Budka, H. Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin. Neuropathol. 2011, 30, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Chinnathambi, S.; Lee, J.J.; Dombroski, B.A.; Baker, M.C.; Soto-Ortolaza, A.I.; Lee, S.E.; Klein, E.; Huang, A.Y.; Sears, R.; et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet. 2012, 21, 3500–3512. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Pastor, P.; Cooper, B.; Cervantes, S.; Benitez, B.A.; Razquin, C.; Goate, A.; Cruchaga, C.; Ibero-American Alzheimer Disease Genetics Group Researchers. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther. 2012, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Ling, H.; Pittman, A.M.; Shaw, K.; de Silva, R.; Simone, R.; Holton, J.L.; Warren, J.D.; Rohrer, J.D.; Xiromerisiou, G.; et al. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol. Aging 2012, 33, 2231.e7–2231.e14. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.Q.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E.M. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Lee, S.E.; Wojta, K.; Ramos, E.M.; Klein, E.; Chen, J.; Boxer, A.L.; Gorno-Tempini, M.L.; Geschwind, D.H.; Schlotawa, L.; et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 2017, 140, 1128–1146. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; Nguyen, T.; Corcoran, N.M.; Velakoulis, D.; Chen, T.; Grundy, R.; O’Brien, T.J.; Hovens, C.M. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol. Dis. 2012, 45, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Shultz, S.R.; Wright, D.K.; Zheng, P.; Stuchbery, R.; Liu, S.J.; Sashindranath, M.; Medcalf, R.L.; Johnston, L.A.; Hovens, C.M.; Jones, N.C.; et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 2015, 138, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Shultz, S.R.; Hovens, C.M.; Velakoulis, D.; Jones, N.C.; O’Brien, T.J. Hyperphosphorylated tau is implicated in acquired epilepsy and neuropsychiatric comorbidities. Mol. Neurobiol. 2014, 49, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.R. The legacy of patient H.M. for neuroscience. Neuron 2009, 61, 6–9. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.P.; Jensen, K.F. Enhanced expression of glial fibrillary acidic protein and the cupric silver degeneration reaction can be used as sensitive and early indicators of neurotoxicity. Neurotoxicology 1992, 13, 113–122. [Google Scholar] [PubMed]


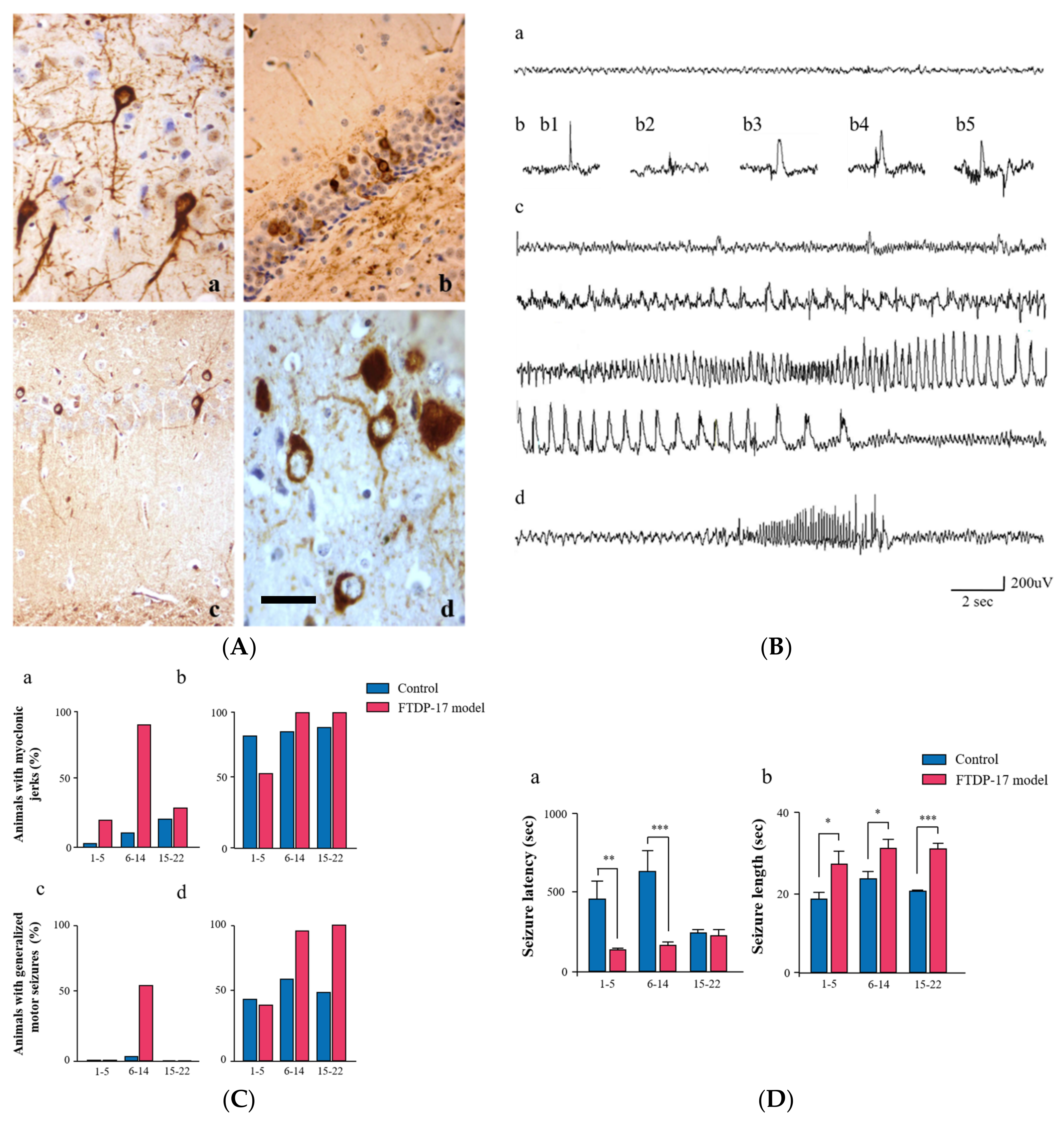{kind=link}
| Diseases | β-Amyloid | Phosopho-tau | α-Synuclein | Epilepsy | Myclonus |
|---|---|---|---|---|---|
| Alzhemier disease (AD) | Yes | Yes | No | Yes | Yes |
| Other dementias: | |||||
| Dementia with Lewy bodies (LBD) | Yes | Yes | Yes | Yes | Yes |
| Frontotemporal dementia (FTD) | No | Yes | No | Yes | Yes |
| Progressive supranuclear palsy (PSP) | No | Yes | No | Yes | Yes |
| Corticobasal degeneration (CBD) | No | Yes | No | No | Yes |
| Down síndrome (DS) | Yes | Yes | No | Yes | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, M.P.; García-Cabrero, A.M.; Sánchez-Elexpuru, G.; Burgos, D.F.; Serratosa, J.M. Tau-Induced Pathology in Epilepsy and Dementia: Notions from Patients and Animal Models. Int. J. Mol. Sci. 2018, 19, 1092. https://doi.org/10.3390/ijms19041092
Sánchez MP, García-Cabrero AM, Sánchez-Elexpuru G, Burgos DF, Serratosa JM. Tau-Induced Pathology in Epilepsy and Dementia: Notions from Patients and Animal Models. International Journal of Molecular Sciences. 2018; 19(4):1092. https://doi.org/10.3390/ijms19041092
Chicago/Turabian StyleSánchez, Marina P., Ana M. García-Cabrero, Gentzane Sánchez-Elexpuru, Daniel F. Burgos, and José M. Serratosa. 2018. "Tau-Induced Pathology in Epilepsy and Dementia: Notions from Patients and Animal Models" International Journal of Molecular Sciences 19, no. 4: 1092. https://doi.org/10.3390/ijms19041092