Assessment of Ab Initio and Density Functional Theory Methods for the Excitations of Donor-Acceptor Complexes: The Case of the Benzene-Tetracyanoethylene Model

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Exchange-Correlation Energy in the Tested DFT Functionals
2.2. Singlet Excitations
2.3. Triplet Excitations
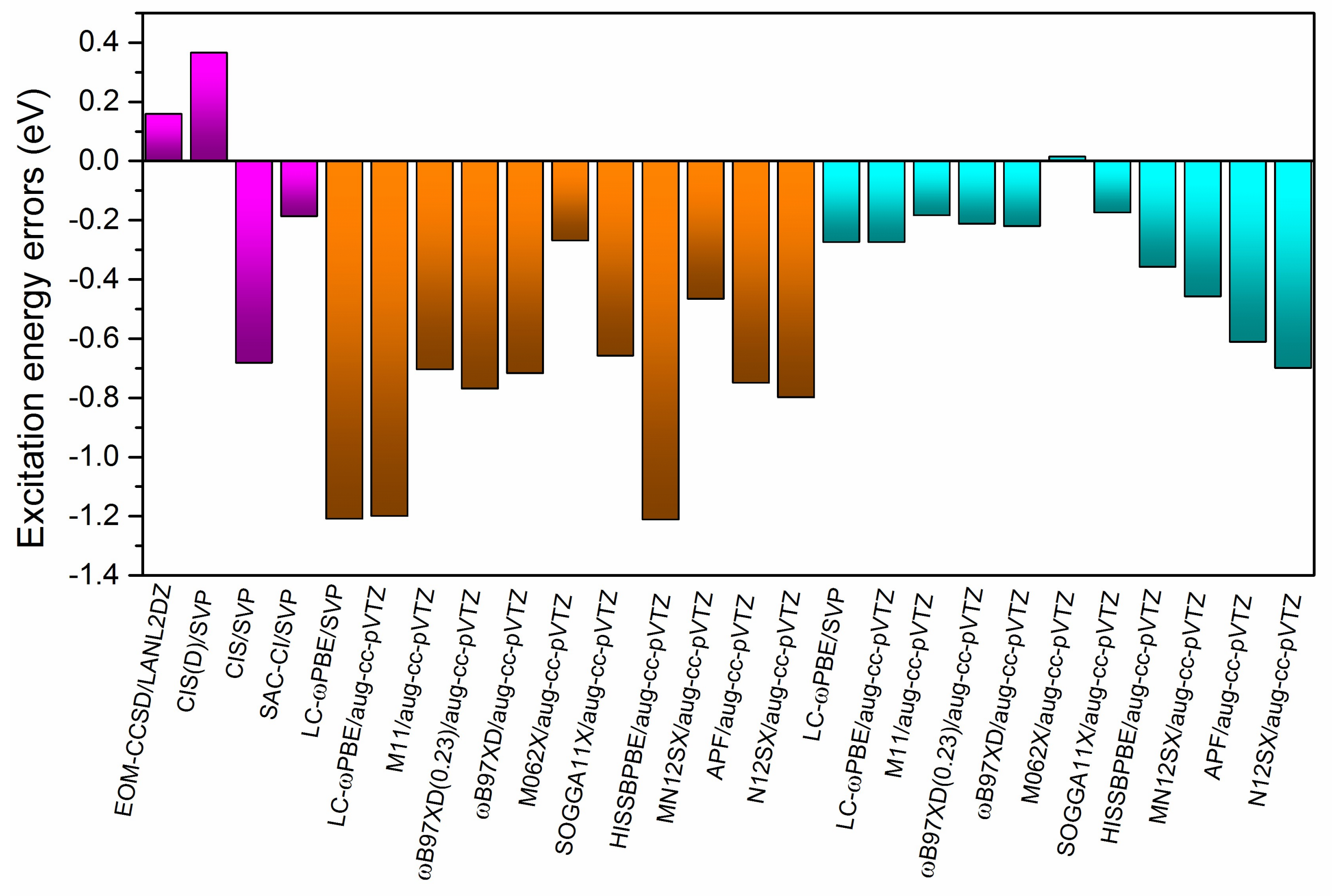
2.4. Assessment of Different Methods for Excited States
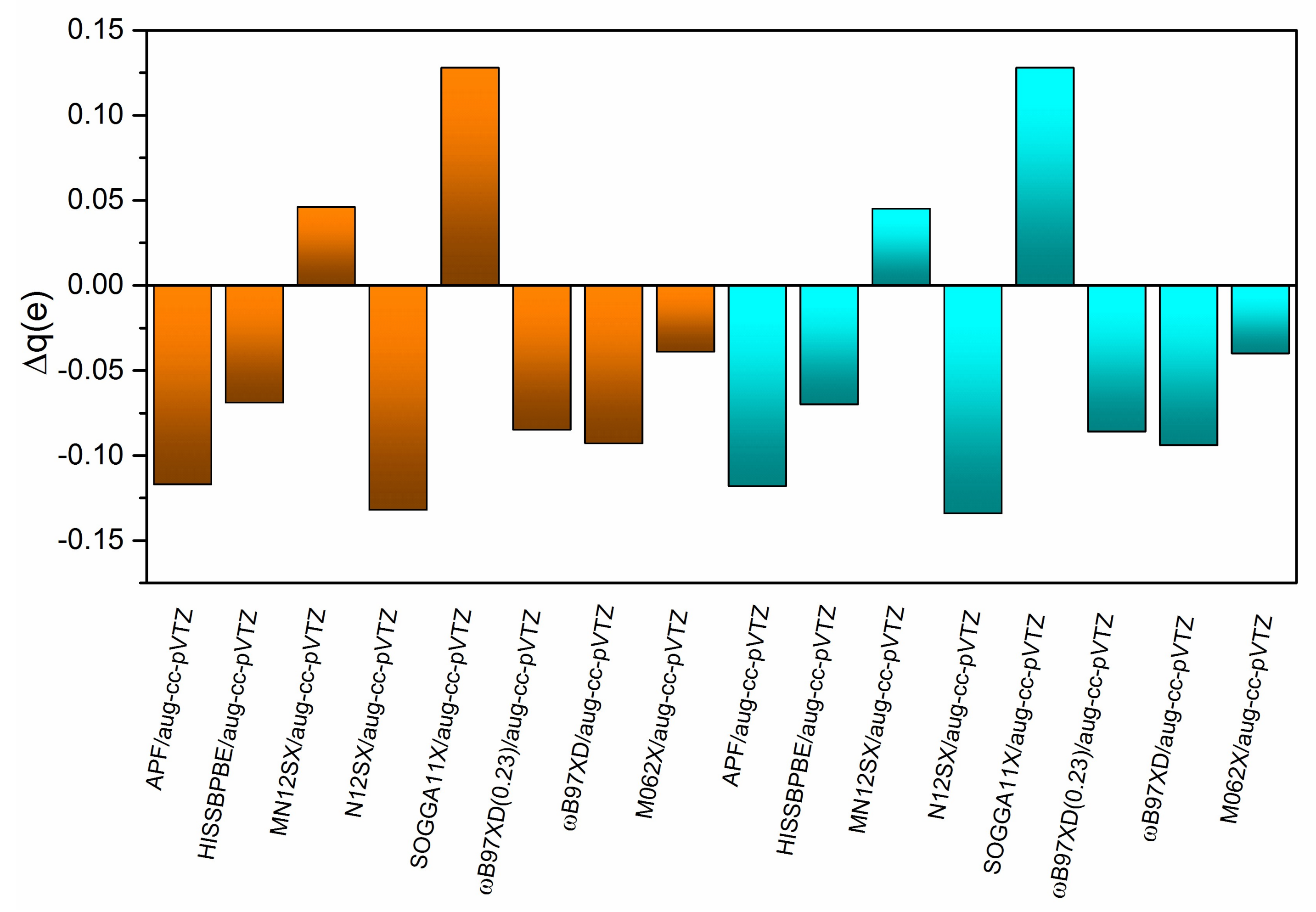
2.5. Transferred Charges from TDDFT
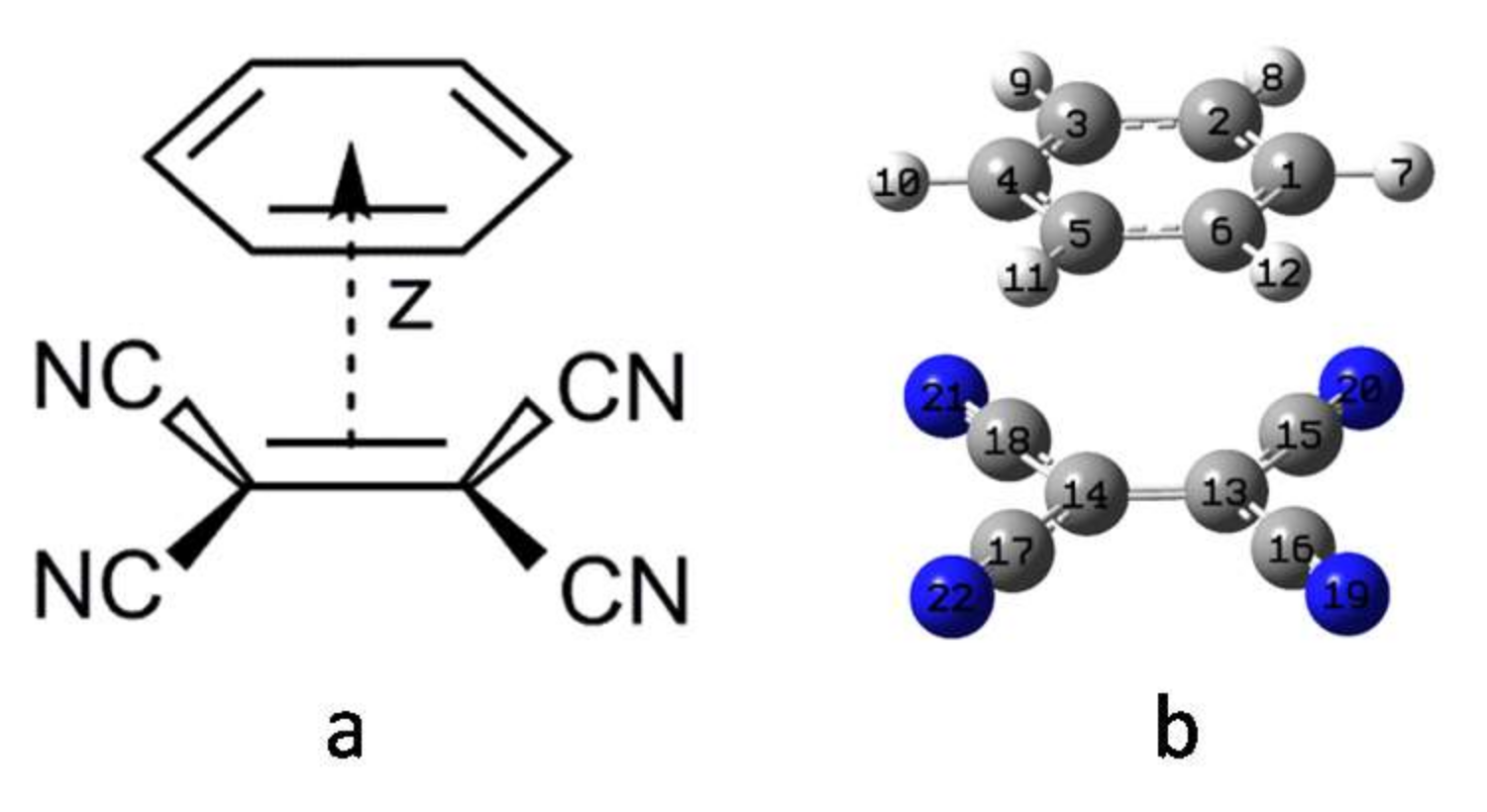
3. Computational Method
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sygletou, M.; Petridis, C.; Kymakis, E.; Stratakis, E. Advanced Photonic Processes for Photovoltaic and Energy Storage Systems. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Bredas, J.L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-transfer and energy-transfer processes in pi-conjugated oligomers and polymers: A molecular picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef] [PubMed]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Bredas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Bredas, J.L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular Understanding of Organic Solar Cells: The Challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Vandewal, K.; Albrecht, S.; Hoke, E.T.; Graham, K.R.; Widmer, J.; Douglas, J.D.; Schubert, M.; Mateker, W.R.; Bloking, J.T.; Burkhard, G.F.; et al. Efficient charge generation by relaxed charge-transfer states at organic interfaces. Nat. Mater. 2014, 13, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, S.; Rao, A.; Kumar, A.; Smith, S.L.; Chin, A.W.; Clark, J.; van der Poll, T.S.; Bazan, G.C.; Friend, R.H. Ultrafast Long-Range Charge Separation in Organic Semiconductor Photovoltaic Diodes. Science 2014, 343, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Beljonne, D.; Cornil, J.; Muccioli, L.; Zannoni, C.; Bredas, J.L.; Castet, F. Electronic Processes at Organic-Organic Interfaces: Insight from Modeling and Implications for Opto-electronic Devices. Chem. Mater. 2011, 23, 591–609. [Google Scholar] [CrossRef]
- Zang, H.; Ke, Y.L.; Zhao, Y.; Liang, W.Z. Effects of Charge Transfer State and Exciton Migration on Singlet Fission Dynamics in Organic Aggregates. J. Phys. Chem. C 2016, 120, 13351–13359. [Google Scholar] [CrossRef]
- Zhu, Z.; Zang, H.; Zhao, Y.; Liang, W. Charge Carrier Mobilities and Singlet Fission Dynamics in Thienoquinoidal Compounds. J. Phys. Chem. C 2017, 121, 22587–22596. [Google Scholar] [CrossRef]
- Zang, H.; Zhao, Y.; Liang, W. Quantum Interference in Singlet Fission: J- and H-Aggregate Behavior. J. Phys. Chem. Lett. 2017, 8, 5105–5112. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, B.; Sun, Z.; Sun, H. Prediction of excited-state properties of oligoacene crystals using polarizable continuum model-tuned range-separated hybrid functional approach. J. Comput. Chem. 2017, 38, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, C.-R.; Yuan, L.-H.; Zhang, M.-L.; Chen, Y.-H.; Liu, Z.-J.; Chen, H.-S. The electronic structures and optical properties of fullerene derivatives for organic solar cells: The number and size effects of fullerene-cage. Mater. Chem. Phys. 2018, 204, 95–104. [Google Scholar] [CrossRef]
- Kümmel, S. Charge-Transfer Excitations: A Challenge for Time-Dependent Density Functional Theory That Has Been Met. Adv. Energy Mater. 2017, 7. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, H.C.; Zhang, S.T.; Gao, Y.; Li, B.; Yan, Y.; Hu, Y.S.; Zhao, L.J.; Yang, B. Discrete face-to-face stacking of anthracene inducing high-efficiency excimer fluorescence in solids via a thermally activated phase transition. J. Mater. Chem. C 2017, 5, 10061–10067. [Google Scholar] [CrossRef]
- Klene, M.; Robb, M.A.; Frisch, M.J.; Celani, P. Parallel implementation of the CI-vector evaluation in full CI/CAS-SCF. J. Chem. Phys. 2000, 113, 5653–5665. [Google Scholar] [CrossRef]
- Takahito Nakajima, H.N. Energy gradient method for the ground, excited, ionized, and electron-attached states calculated by the SAC (symmetry-adapted cluster)/SAC-CI (configuration interaction metod). J. Chem. Phys. 1999, 242, 177–193. [Google Scholar]
- Stanton, J.F.; Bartlett, R.J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Rico, R.J.; Oumi, M.; Lee, T.J. A doubles correction to electronic excited states from configuration interaction in the space of single substitutions. Chem. Phys. Lett. 1994, 219, 21–29. [Google Scholar] [CrossRef]
- Hung, L.; Bruneval, F.; Baishya, K.; Ogut, S. Benchmarking the GW Approximation and Bethe-Salpeter Equation for Groups IB and IIB Atoms and Monoxides. J. Chem. Theory Comput. 2017, 13, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Kuritz, N.; Stein, T.; Baer, R.; Kronik, L. Charge-Transfer-Like pi-->pi* Excitations in Time-Dependent Density Functional Theory: A Conundrum and Its Solution. J. Chem. Theory Comput. 2011, 7, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; Fias, S.; Boisdenghien, Z.; De Proft, F. Conceptual DFT: Chemistry from the linear response function. Chem. Soc. Rev. 2014, 43, 4989–5008. [Google Scholar] [CrossRef] [PubMed]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Williamson, M.J.; Tozer, D.J. Influence of Triplet Instabilities in TDDFT. J. Chem. Theory Comput. 2011, 7, 3578–3585. [Google Scholar] [CrossRef] [PubMed]
- Heuser, J.; Hofener, S. Analytical nuclear excited-state gradients for the Tamm-Dancoff approximation using uncoupled frozen-density embedding. J. Comput. Chem. 2017, 38, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Chantzis, A.; Laurent, A.D.; Adamo, C.; Jacquemin, D. Is the Tamm-Dancoff Approximation Reliable for the Calculation of Absorption and Fluorescence Band Shapes? J. Chem. Theory Comput. 2013, 9, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Refaely-Abramson, S.; Baer, R.; Kronik, L. Fundamental and excitation gaps in molecules of relevance for organic photovoltaics from an optimally tuned range-separated hybrid functional. Phys. Rev. B 2011, 84, 9226–9231. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Chem. Phys. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT Study of van der Waals Complexes: Rare-Gas Dimers, Alkaline-Earth Dimers, Zinc Dimer, and Zinc-Rare-Gas Dimers. J. Phys. Chem. A 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Frisch, M.J.; Kudin, K.N.; Schlegel, H.B.; Morokuma, K. Geometry optimization with QM/MM methods II: Explicit quadratic coupling. Mol. Phys. 2006, 104, 701–714. [Google Scholar] [CrossRef]
- Constantin, L.A.; Pitarke, J.M. Simple dynamic exchange-correlation kernel of a uniform electron gas. Phys. Rev. B 2007, 75, 245127. [Google Scholar] [CrossRef]
- Constantin, L.A. Simple effective interaction for dimensional crossover. Phys. Rev. B 2016, 93, 121104. [Google Scholar] [CrossRef]
- Stein, T.; Kronik, L.; Baer, R. Prediction of charge-transfer excitations in coumarin-based dyes using a range-separated functional tuned from first principles. J. Chem. Phys. 2009, 131, 244119. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Autschbach, J.; Govind, N.; Kronik, L.; Baer, R. Curvature and Frontier Orbital Energies in Density Functional Theory. J. Phys. Chem. Lett. 2012, 3, 3740–3744. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.H.; Hu, Z.B.; Sun, Z.R.; Sun, H.T. Density Functional Theory Studies on Ionization Energies, Electron Affinities, and Polarization Energies of Organic Semiconductors. Acta Phys. Chim. Sin. 2017, 33, 1171–1180. [Google Scholar]
- Sun, H.T.; Zhong, C.; Sun, Z.R. Recent Advances in the Optimally “Tuned” Range-Separated Density Functional Theory. Acta Phys. Chim. Sin. 2016, 32, 2197–2208. [Google Scholar]
- Stein, T.; Kronik, L.; Baer, R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-R.; Sears, J.S.; Yang, B.; Aziz, S.G.; Coropceanu, V.; Brédas, J.-L. Theoretical Study of the Local and Charge-Transfer Excitations in Model Complexes of Pentacene-C60 Using Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2014, 10, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yi, Y.; Zhang, C.-R.; Aziz, S.G.; Coropceanu, V.; Brédas, J.-L. Impact of Electron Delocalization on the Nature of the Charge-Transfer States in Model Pentacene/C60 Interfaces: A Density Functional Theory Study. J. Phys. Chem. C 2014, 118, 27648–27656. [Google Scholar] [CrossRef]
- Sutton, C.; Korzdorfer, T.; Gray, M.T.; Brunsfeld, M.; Parrish, R.M.; Sherrill, C.D.; Sears, J.S.; Bredas, J.L. Accurate description of torsion potentials in conjugated polymers using density functionals with reduced self-interaction error. J. Chem. Phys. 2014, 140, 054310. [Google Scholar] [CrossRef] [PubMed]
- Nan, G.; Zhang, X.; Lu, G. The lowest-energy charge-transfer state and its role in charge separation in organic photovoltaics. Phys. Chem. Chem. Phys. 2016, 18, 17546–17556. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-K.; Ravva, M.K.; Li, H.; Ryno, S.M.; Brédas, J.-L. Effect of Molecular Packing and Charge Delocalization on the Nonradiative Recombination of Charge-Transfer States in Organic Solar Cells. Adv. Energy Mater. 2016, 6. [Google Scholar] [CrossRef]
- Rangel, T.; Hamed, S.M.; Bruneval, F.; Neaton, J.B. Evaluating the GW Approximation with CCSD(T) for Charged Excitations Across the Oligoacenes. J. Chem. Theory Comput. 2016, 12, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Van Regemorter, T.; Guillaume, M.; Sini, G.; Sears, J.S.; Geskin, V.; Brédas, J.-L.; Beljonne, D.; Cornil, J. Density functional theory for the description of charge-transfer processes at TTF/TCNQ interfaces. Theor. Chem. Acc. 2012, 131, 1273. [Google Scholar] [CrossRef]
- Van Regemorter, T.; Guillaume, M.; Fuchs, A.; Lennartz, C.; Geskin, V.; Beljonne, D.; Cornil, J. Methodological aspects of the quantum-chemical description of interface dipoles at tetrathiafulvalenetetracyanoquinodimethane interfaces. J. Chem. Phys. 2012, 137, 174708. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Nakano, M.; Castet, F.D.R. Nonempirically Tuned Long-Range Corrected Density Functional Theory Study on Local and Charge-Transfer Excitation Energies in a Pentacene/C60 Model Complex. J. Phys. Chem. Lett. 2011, 2, 1725–1730. [Google Scholar] [CrossRef]
- Minami, T.; Ito, S.; Nakano, M. Functional dependence of excitation energy for pentacene/C60 model complex in the nonempirically tuned long-range corrected density functional theory. Int. J. Quantum Chem. 2013, 113, 252–256. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry. J. Chem. Phys. 2011, 135, 191102. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.M.; Izmaylov, A.F.; Scuseria, G.E.; Savin, A. Assessment of a Middle-Range Hybrid Functional. J. Chem. Theory Comput. 2008, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Harding, M.E.; Seiler, C.; Weigend, F.; Evers, F.; van Setten, M.J. Quasi-Particle Self-Consistent GW for Molecules. J. Chem. Theory Comput. 2016, 12, 2528–2541. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C. Accurate Simulation of Optical Properties in Dyes. Acc. Chem. Res. 2009, 42, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Harms, J.; Caricato, M. An EOM-CCSD-PCM Benchmark for Electronic Excitation Energies of Solvated Molecules. J. Chem. Theory Comput. 2017, 13, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Chaudhuri, S.; Batista, V.S. Can TDDFT Describe Excited Electronic States of Naphthol Photoacids? A Closer Look with EOM-CCSD. J. Chem. Theory Comput. 2018, 14, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- Sini, G.; Sears, J.S.; Bredas, J.L. Evaluating the Performance of DFT Functionals in Assessing the Interaction Energy and Ground-State Charge Transfer of Donor/Acceptor Complexes: Tetrathiafulvalene-Tetracyanoquinodimethane (TTF-TCNQ) as a Model Case. J. Chem. Theory Comput. 2011, 7, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Duchemin, I.; Blondel, A.; Blase, X. Benchmark of Bethe-Salpeter for Triplet Excited-States. J. Chem. Theory Comput. 2017, 13, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.L.; Yang, W.T. Challenges with range-separated exchange-correlation functionals in time-dependent density functional theory calculations. Mol. Phys. 2010, 108, 2745–2750. [Google Scholar] [CrossRef]
- Mewes, S.A.; Plasser, F.; Krylov, A.; Dreuw, A. Benchmarking excited-state calculations using exciton properties. J. Chem. Theory Comput. 2018, 14, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Liao, M.-S.; Lu, Y.; Parker, V.D.; Scheiner, S. DFT Calculations and Spectral Measurements of Charge-Transfer Complexes Formed by Aromatic Amines and Nitrogen Heterocycles with Tetracyanoethylene and Chloranil. J. Phys. Chem. A 2003, 107, 8939–8948. [Google Scholar] [CrossRef]
- Arago, J.; Sancho-Garcia, J.C.; Orti, E.; Beljonne, D. Ab Initio Modeling of Donor-Acceptor Interactions and Charge-Transfer Excitations in Molecular Complexes: The Case of Terthiophene-Tetracyanoquinodimethane. J. Chem. Theory Comput. 2011, 7, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F. Calculation of Molecular Orbital Composition. Acta Chim. Sin. 2011, 69, 2393–2406. [Google Scholar]
- Li, X.-Y.; Zhang, C.-R.; Yuan, L.-H.; Zhang, M.-L.; Chen, Y.-H.; Liu, Z.-J. A comparative study of porphyrin dye sensitizers YD2-o-C8, SM315 and SM371 for solar cells: The electronic structures and excitation-related properties. Eur. Phys. J. D 2016, 70, 211. [Google Scholar] [CrossRef]
- Li, S.-H.; Zhang, C.-R.; Yuan, L.-H.; Zhang, M.-L.; Chen, Y.-H.; Liu, Z.-J.; Chen, H.-S. The role of electronic donor moieties in porphyrin dye sensitizers for solar cells: Electronic structures and excitation related properties. J. Renew. Sustain. Energy 2017, 9, 053505. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFT Functionals | Key Parameters | |
|---|---|---|
| APF | ||
| SOGGA11X | x = 40.15 | |
| LC-ωPBE | ω = 0.4 a0−1 | |
| ωB97XD | ω = 0.2 a0−1 cx = 0.222 | |
| N12SX/MN12SX | x = 25 ω = 0.11 a0−1 | |
| HISSbPBE | = 0.84 a0−1 = 0.20 a0−1 | |
| M06-2X | x = 54 | |
| M11 | x = 42.8 ω = 0.25 a0−1 |
| Excited States | Main Transition Configurations | Excitation Energies (eV/nm) |
|---|---|---|
| S1 | H→L (90.3%) | 4.19/296 |
| S2 | H−1→L (90.2%) | 4.23/293 |
| S3 | H−1→L+2 (42.9%); H→L+3 (31.2%); H→L+1 (13.4%) | 5.34/232 |
| T1 | H−2→L (86.1%) | 2.71/457 |
| T2 | H→L+2 (44.8%); H−1→L+3 (32.0%); H−1→L+1 (12.4%) | 4.08/304 |
| T3 | H→L (89.5%) | 4.16/297 |
| Method | S-CT1 (eV) | S-LE1 (eV) | T1 (eV) |
|---|---|---|---|
| EOM-CCSD/LanL2DZ | 4.4673 | 5.3663 | 2.8692 |
| EOM-CCSD/SVP | 4.1882 | 5.3402 | 2.7103 |
| CIS(D)/SVP | 3.4807 | 5.5586 | 3.0764 |
| CIS/SVP | 4.6855 | 5.2257 | 2.0291 |
| SAC-CI/SVP | 3.9641 | 5.4669 | 2.5240 |
| LC-ωPBE/SVP | 4.0198 | 4.8595 | 1.5016 |
| LC-ωPBE/aug-cc-pVTZ | 3.9090 | 4.7404 | 1.5110 |
| M11/aug-cc-pVTZ | 3.5738 | 4.6509 | 2.0067 |
| ωB97XD(0.23)/aug-cc-pVTZ | 3.2486 | 4.5466 | 1.9417 |
| ωB97XD/aug-cc-pVTZ | 3.0399 | 4.4959 | 1.9934 |
| M062X/aug-cc-pVTZ | 3.0142 | 4.5908 | 2.4418 |
| SOGGA11X/aug-cc-pVTZ | 2.7162 | 4.4993 | 2.0527 |
| HISSbPBE/aug-cc-pVTZ | 2.4667 | 4.5193 | 1.4994 |
| MN12SX/aug-cc-pVTZ | 2.2881 | 4.3012 | 2.2448 |
| APF/aug-cc-pVTZ | 2.1206 | 4.3254 | 1.9606 |
| N12SX/aug-cc-pVTZ | 2.0305 | 4.2905 | 1.9121 |
| (TDA) LC-ωPBE/SVP | 4.0263 | 5.2205 | 2.4361 |
| (TDA)LC-ωPBE/aug-cc-pVTZ | 3.9160 | 5.0828 | 2.4365 |
| (TDA)M11/aug-cc-pVTZ | 3.5799 | 5.0313 | 2.5272 |
| (TDA)ωB97XD(0.23)/aug-cc-pVTZ | 3.2547 | 4.8925 | 2.4985 |
| (TDA)ωB97XD/aug-cc-pVTZ | 3.0455 | 4.8483 | 2.4905 |
| (TDA)M062X/aug-cc-pVTZ | 3.0191 | 4.9585 | 2.7250 |
| (TDA)SOGGA11X/aug-cc-pVTZ | 2.7205 | 4.8491 | 2.5366 |
| (TDA)HISSbPBE/aug-cc-pVTZ | 2.4703 | 4.8803 | 2.3527 |
| (TDA)MN12SX/aug-cc-pVTZ | 2.2915 | 4.6294 | 2.2528 |
| (TDA)APF/aug-cc-pVTZ | 2.1242 | 4.6105 | 2.0993 |
| (TDA)N12SX/aug-cc-pVTZ | 2.0340 | 4.5779 | 2.0113 |
| Method | AMAE (eV) | SE (eV) |
|---|---|---|
| EOM-CCSD/LanL2DZ | 0.1547 | 0.1860 |
| CIS(D)/SVP | 0.4306 | 0.4769 |
| CIS/SVP | 0.4310 | 0.4914 |
| SAC-CI/SVP | 0.1790 | 0.1835 |
| LC-ωPBE/SVP | 0.6193 | 0.7573 |
| LC-ωPBE/aug-cc-pVTZ | 0.6928 | 0.7908 |
| M11/aug-cc-pVTZ | 0.6691 | 0.6702 |
| ωB97XD(0.23)/aug-cc-pVTZ | 0.8339 | 0.8373 |
| ωB97XD/aug-cc-pVTZ | 0.9032 | 0.9211 |
| M062X/aug-cc-pVTZ | 0.7306 | 0.8189 |
| SOGGA11X/aug-cc-pVTZ | 0.9902 | 1.0498 |
| HISSbPBE/aug-cc-pVTZ | 1.2511 | 1.3043 |
| MN12SX/aug-cc-pVTZ | 1.1349 | 1.2789 |
| APF/aug-cc-pVTZ | 1.2779 | 1.3984 |
| N12SX/aug-cc-pVTZ | 1.3352 | 1.4600 |
| (TDA) LC-ωPBE/SVP | 0.1853 | 0.1964 |
| (TDA) LC-ωPBE/aug-cc-pVTZ | 0.2678 | 0.2679 |
| (TDA) M11/aug-cc-pVTZ | 0.3668 | 0.4078 |
| (TDA) ωB97XD(0.23)/aug-cc-pVTZ | 0.5310 | 0.6101 |
| (TDA) ωB97XD/aug-cc-pVTZ | 0.6181 | 0.7294 |
| (TDA) M062X/aug-cc-pVTZ | 0.5218 | 0.7101 |
| (TDA) SOGGA11X/aug-cc-pVTZ | 0.7108 | 0.8992 |
| (TDA) HISSbPBE/aug-cc-pVTZ | 0.8451 | 1.0473 |
| (TDA) MN12SX/aug-cc-pVTZ | 1.0217 | 1.1989 |
| (TDA) APF/aug-cc-pVTZ | 1.1349 | 1.3122 |
| (TDA) N12SX/aug-cc-pVTZ | 1.2052 | 1.3796 |
| Methods | ||
|---|---|---|
| APF | 1.240 | 2.079 |
| HISSbPBE | 1.288 | 2.045 |
| M11 | 1.357 | 2.012 |
| MN12SX | 1.403 | 1.848 |
| N12SX | 1.225 | 2.101 |
| SOGGA11X | 1.485 | 1.784 |
| ωB97XD(0.23) | 1.272 | 2.130 |
| ωB97XD | 1.264 | 2.135 |
| M062X | 1.318 | 2.028 |
| (TDA) APF | 1.239 | 2.078 |
| (TDA) HISSbPBE | 1.287 | 2.045 |
| (TDA) M11 | 1.357 | 2.012 |
| (TDA) MN12SX | 1.402 | 1.847 |
| (TDA) N12SX | 1.223 | 2.100 |
| (TDA) SOGGA11X | 1.485 | 1.783 |
| (TDA) ωB97XD(0.23) | 1.271 | 2.130 |
| (TDA)ωB97XD | 1.263 | 2.135 |
| (TDA) M062X | 1.317 | 2.028 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Zhang, C.-R.; Wang, W.; Gong, J.-J.; Liu, Z.-J.; Chen, H.-S. Assessment of Ab Initio and Density Functional Theory Methods for the Excitations of Donor-Acceptor Complexes: The Case of the Benzene-Tetracyanoethylene Model. Int. J. Mol. Sci. 2018, 19, 1134. https://doi.org/10.3390/ijms19041134
Xu P, Zhang C-R, Wang W, Gong J-J, Liu Z-J, Chen H-S. Assessment of Ab Initio and Density Functional Theory Methods for the Excitations of Donor-Acceptor Complexes: The Case of the Benzene-Tetracyanoethylene Model. International Journal of Molecular Sciences. 2018; 19(4):1134. https://doi.org/10.3390/ijms19041134
Chicago/Turabian StyleXu, Peng, Cai-Rong Zhang, Wei Wang, Ji-Jun Gong, Zi-Jiang Liu, and Hong-Shan Chen. 2018. "Assessment of Ab Initio and Density Functional Theory Methods for the Excitations of Donor-Acceptor Complexes: The Case of the Benzene-Tetracyanoethylene Model" International Journal of Molecular Sciences 19, no. 4: 1134. https://doi.org/10.3390/ijms19041134
APA StyleXu, P., Zhang, C.-R., Wang, W., Gong, J.-J., Liu, Z.-J., & Chen, H.-S. (2018). Assessment of Ab Initio and Density Functional Theory Methods for the Excitations of Donor-Acceptor Complexes: The Case of the Benzene-Tetracyanoethylene Model. International Journal of Molecular Sciences, 19(4), 1134. https://doi.org/10.3390/ijms19041134