Defining Driver DNA Methylation Changes in Human Cancer

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
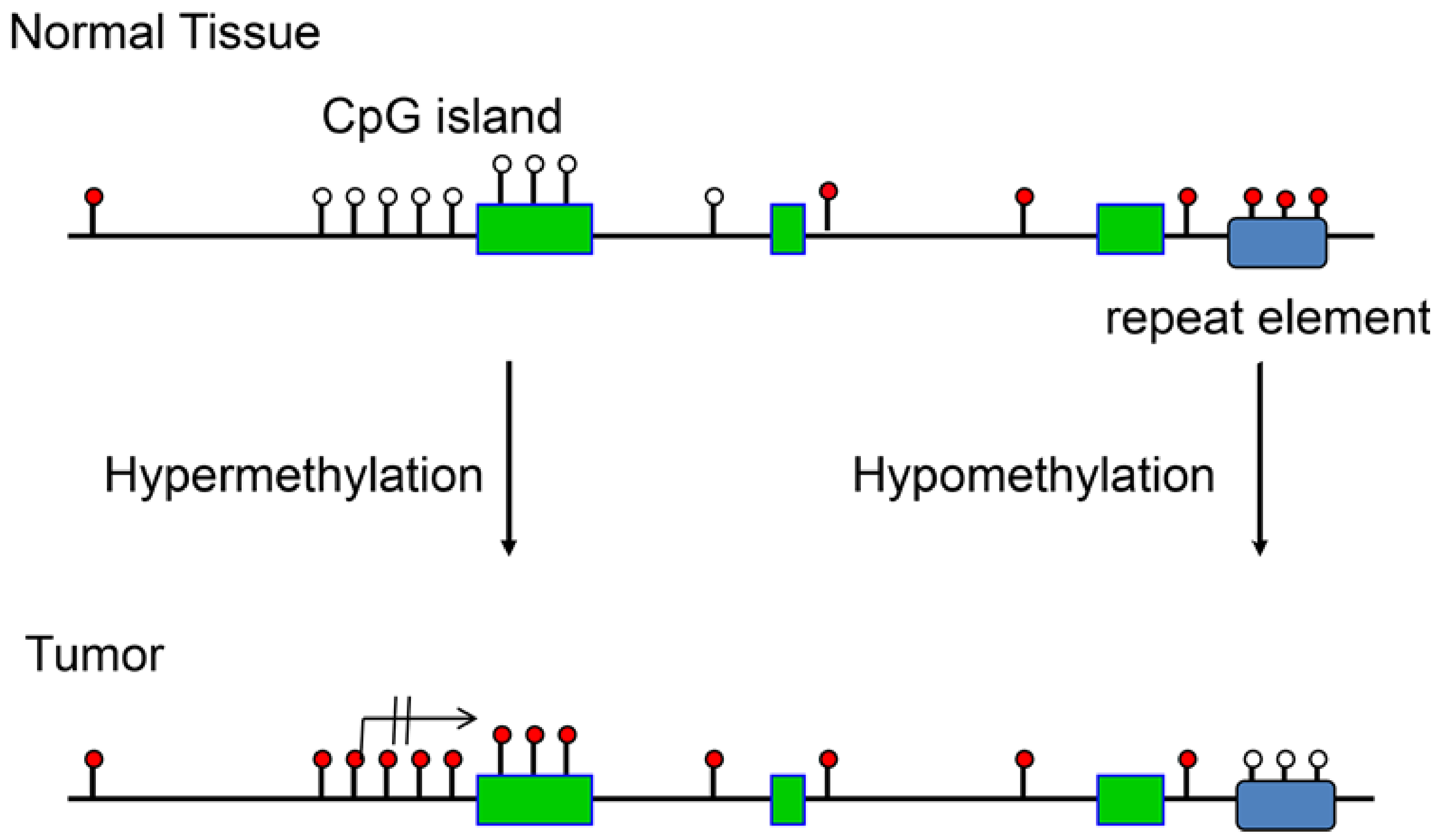
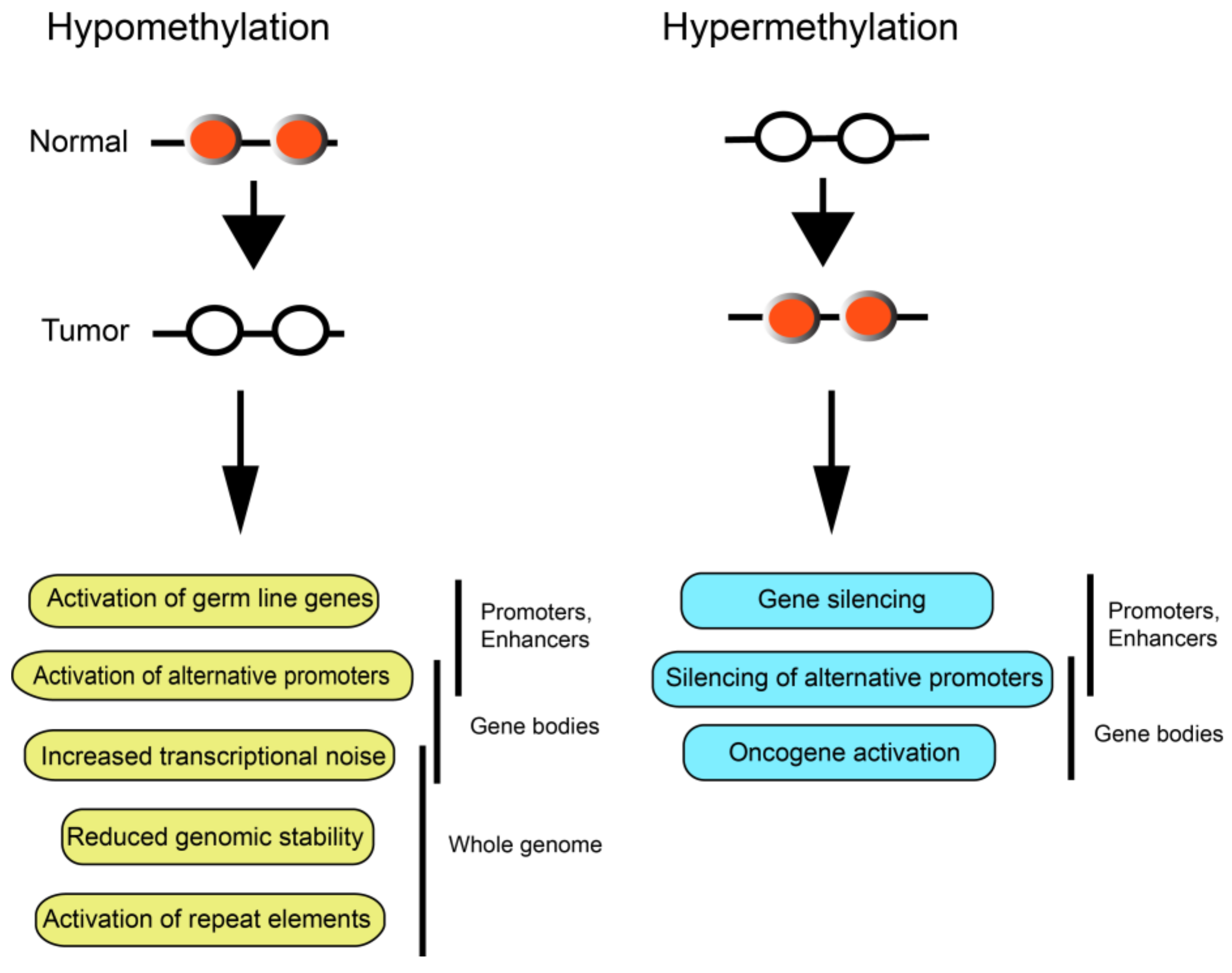
2. DNA Hypomethylation during Tumor Formation
3. Tumor-Driving DNA Hypermethylation Events
4. Classical Tumor Suppressor Genes (TSGs)—BRCA1, MLH1, APC, and CDKN2A
5. Other Likely Tumor Suppressor Genes: DNA Repair Genes, Proapoptotic Genes, and Anti-Proliferative Factors Functioning in Signaling Pathways
6. Transcription Factors Involved in Differentiation Pathways
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 5mC | 5-methylcytosine |
| APC | adenomatous polyposis gene |
| BRCA1 | breast cancer 1 gene |
| CDKN2A | cyclin-dependent kinase inhibitor 2A gene |
| DAPK | death associated protein kinase |
| DNMT | DNA methyltransferase enzyme |
| LINE | long interspersed nuclear element |
| MAGE | melanoma antigen gene |
| MGMT | O6-methylguanine methyltransferase |
| MLH1 | MutL homologue 1 |
| RASSF1A | ras association family 1A |
| SFRP | secreted frizzled-related protein |
| TET | Ten-Eleven-Translocation enzyme |
References
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenet. Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Guibert, S.; Forne, T.; Weber, M. Dynamic regulation of DNA methylation during mammalian development. Epigenomics 2009, 1, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Lacey, M. DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 2013, 754, 31–56. [Google Scholar] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- Romanov, G.A.; Vanyushin, B.F. Methylation of reiterated sequences in mammalian DNAs. Effects of the tissue type, age, malignancy and hormonal induction. Biochim. Biophys. Acta 1981, 653, 204–218. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 1983, 111, 47–54. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Hoppener, J.W.; de Bustros, A.; Steenbergh, P.H.; Lips, C.J.; Nelkin, B.D. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986, 46, 2917–2922. [Google Scholar] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Mutagenesis at methylated CpG sequences. Curr. Top. Microbiol. Immunol. 2006, 301, 259–281. [Google Scholar] [PubMed]
- Pfeifer, G.P. DNA methylation and mutation. Encycl. Life Sci. Wiley 2017. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.Q.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro Oncol. 2017, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.A.; Zhong, X.; Wu, X.; Wang, M.; Kernstine, K.H.; Wang, Z.; Riggs, A.D.; Pfeifer, G.P. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.; Li, H.; Wu, X.; Pfeifer, G.P. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006, 66, 7939–7947. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.; Wang, Z.; Zhang, X.; Zhong, X.; Wu, X.; Lau, S.K.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc. Natl. Acad. Sci. USA 2007, 104, 5527–5532. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Li, A.X.; Tran, D.A.; Oates, N.; Kang, E.R.; Wu, X.; Szabo, P.E. De novo DNA methylation in the male germ line occurs by default but is excluded at sites of H3K4 methylation. Cell Rep. 2013, 4, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Kalari, S.; Pfeifer, G.P. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet. 2010, 70, 277–308. [Google Scholar] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Omar, W.A.; Tomaszewski, B.; De Prins, S.; Jacobs, G.; Koppen, G.; Mathers, J.C.; Langie, S.A. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS ONE 2013, 8, e79044. [Google Scholar] [CrossRef] [PubMed]
- Tubio, J.M.C.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed]
- De Smet, C.; Loriot, A.; Boon, T. Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Mol. Cell. Biol. 2004, 24, 4781–4790. [Google Scholar] [CrossRef] [PubMed]
- Van Tongelen, A.; Loriot, A.; De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017, 396, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, Y.; Zhou, Y.; Sun, Y.; Sun, W.; Wang, L.; Zhou, C.; Zhou, J.; Zhang, J. Identification of a novel human cancer/testis gene MAEL that is regulated by DNA methylation. Mol. Biol. Rep. 2010, 37, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dai, Y.; Chen, J.; Zeng, T.; Li, Y.; Chen, L.; Zhu, Y.H.; Li, J.; Li, Y.; Ma, S.; et al. Maelstrom promotes hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition by way of Akt/GSK-3β/Snail signaling. Hepatology 2014, 59, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Ladelfa, M.F.; Peche, L.Y.; Toledo, M.F.; Laiseca, J.E.; Schneider, C.; Monte, M. Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 2012, 325, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Alberti, L.; Vostrov, A.; Kwon, Y.W.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. BORIS/CTCFL-mediated transcriptional regulation of the hTERT telomerase gene in testicular and ovarian tumor cells. Nucleic Acids Res. 2011, 39, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.A.; Wu, X.; Zhong, X.; Riggs, A.D.; Pfeifer, G.P. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Hanley, M.P.; Hahn, M.A.; Li, A.X.; Wu, X.; Lin, J.; Wang, J.; Choi, A.H.; Ouyang, Z.; Fong, Y.; Pfeifer, G.P.; et al. Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 2017, 36, 5035–5044. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Lucena-Porcel, C.; Kohler, F.; Wurzer, E.; Schmidt, K.; Gallinat, S.; Wenck, H.; et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat. Commun. 2018, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Weisenberger, D.J. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 2017, 12, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350. [Google Scholar] [PubMed]
- Catteau, A.; Harris, W.H.; Xu, C.F.; Solomon, E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: Correlation with disease characteristics. Oncogene 1999, 18, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar] [PubMed]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar] [PubMed]
- Liang, T.J.; Wang, H.X.; Zheng, Y.Y.; Cao, Y.Q.; Wu, X.; Zhou, X.; Dong, S.X. APC hypermethylation for early diagnosis of colorectal cancer: A meta-analysis and literature review. Oncotarget 2017, 8, 46468–46479. [Google Scholar] [CrossRef] [PubMed]
- Lahtz, C.; Pfeifer, G.P. Epigenetic changes of DNA repair genes in cancer. J. Mol. Cell Biol. 2011, 3, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar] [PubMed]
- Paz, M.F.; Yaya-Tur, R.; Rojas-Marcos, I.; Reynes, G.; Pollan, M.; Aguirre-Cruz, L.; Garcia-Lopez, J.L.; Piquer, J.; Safont, M.J.; Balana, C.; et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin. Cancer Res. 2004, 10, 4933–4938. [Google Scholar] [CrossRef] [PubMed]
- Zochbauer-Muller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar] [PubMed]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.G.; Xiong, W.; Wu, X.; Yang, L.; Pfeifer, G.P. The DNA methylation landscape of human melanoma. Genomics 2015, 106, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Kiss, N.B.; Kogner, P.; Johnsen, J.I.; Martinsson, T.; Larsson, C.; Geli, J. Quantitative global and gene-specific promoter methylation in relation to biological properties of neuroblastomas. BMC Med. Genet. 2012, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Krug, U.; Ganser, A.; Koeffler, H.P. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 2002, 21, 3475–3495. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schagdarsurengin, U.; Blumke, K.; Wurl, P.; Pfeifer, G.P.; Hauptmann, S.; Taubert, H.; Dammann, R. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol. Carcinog. 2007, 46, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [PubMed]
- Dammann, R.; Schagdarsurengin, U.; Seidel, C.; Strunnikova, M.; Rastetter, M.; Baier, K.; Pfeifer, G.P. The tumor suppressor RASSF1A in human carcinogenesis: An update. Histol. Histopathol. 2005, 20, 645–663. [Google Scholar] [PubMed]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.K.; Dammann, R.; Pfeifer, G.P. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Khokhlatchev, A.; Rabizadeh, S.; Xavier, R.; Nedwidek, M.; Chen, T.; Zhang, X.F.; Seed, B.; Avruch, J. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 2002, 12, 253–265. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rauch, T.A.; Zhong, X.; Bennett, W.P.; Latif, F.; Krex, D.; Pfeifer, G.P. CpG island hypermethylation in human astrocytomas. Cancer Res. 2010, 70, 2718–2727. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. https://doi.org/10.3390/ijms19041166
Pfeifer GP. Defining Driver DNA Methylation Changes in Human Cancer. International Journal of Molecular Sciences. 2018; 19(4):1166. https://doi.org/10.3390/ijms19041166
Chicago/Turabian StylePfeifer, Gerd P. 2018. "Defining Driver DNA Methylation Changes in Human Cancer" International Journal of Molecular Sciences 19, no. 4: 1166. https://doi.org/10.3390/ijms19041166
APA StylePfeifer, G. P. (2018). Defining Driver DNA Methylation Changes in Human Cancer. International Journal of Molecular Sciences, 19(4), 1166. https://doi.org/10.3390/ijms19041166