Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
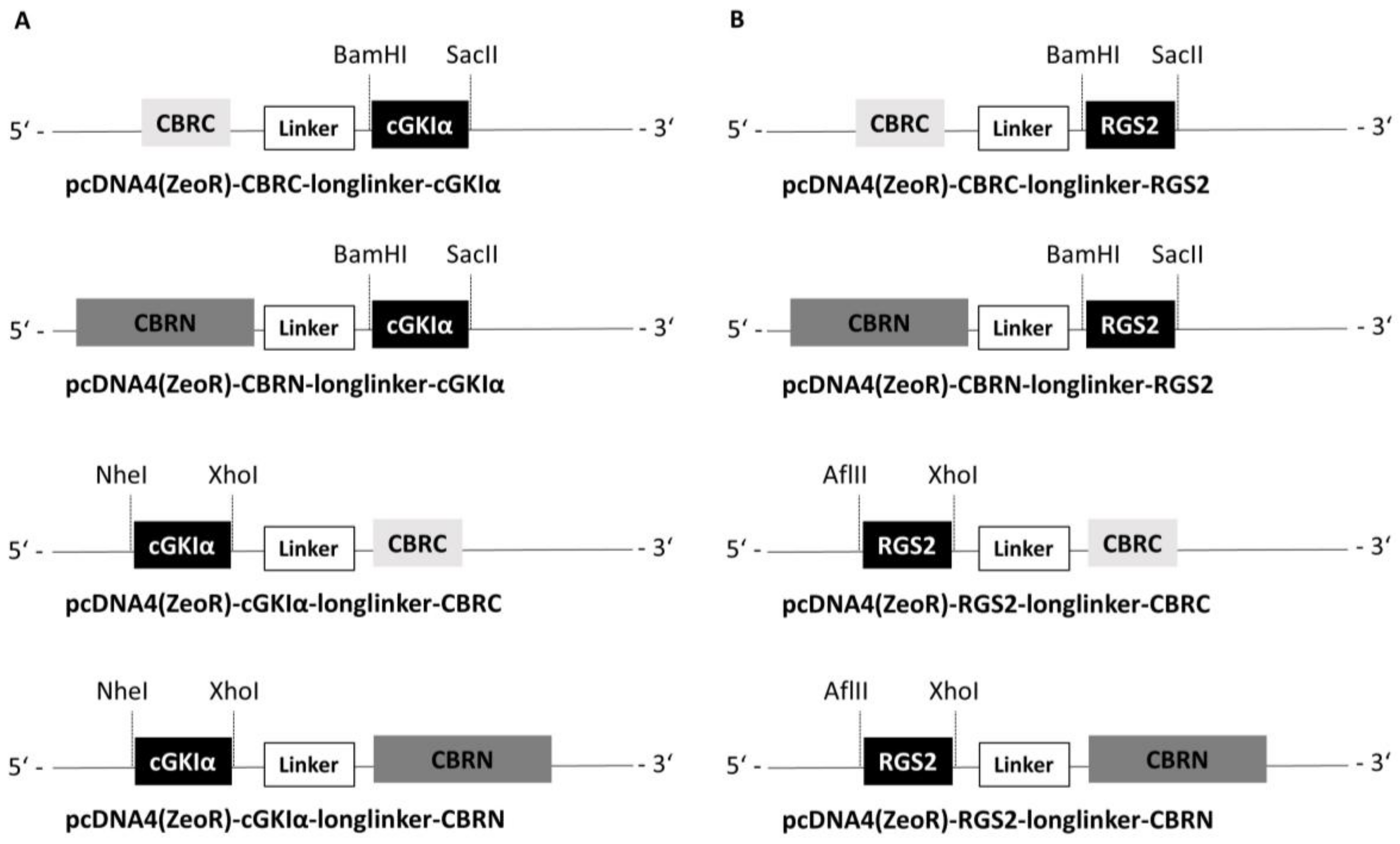
2.1. Construction of Vectors
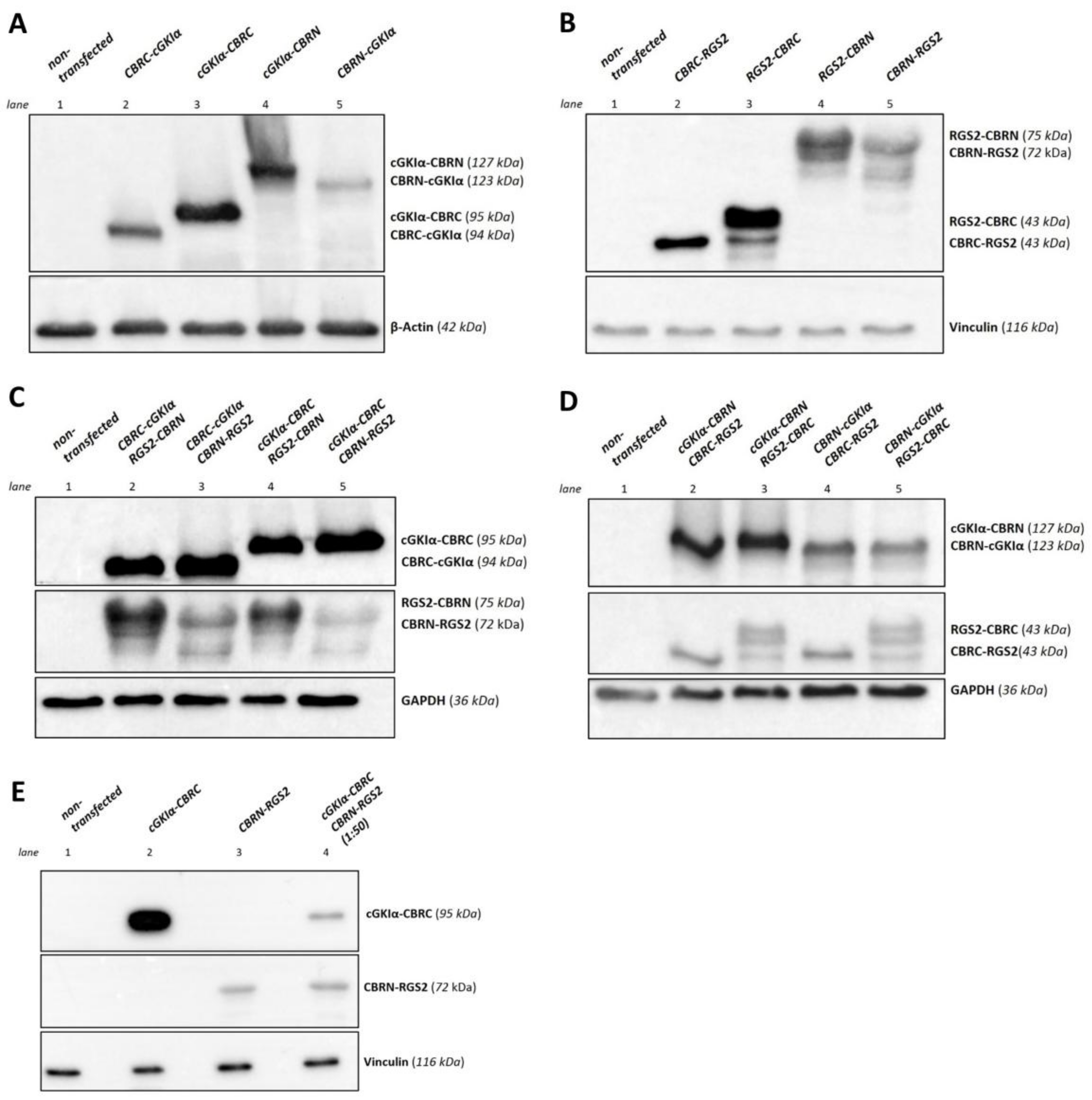
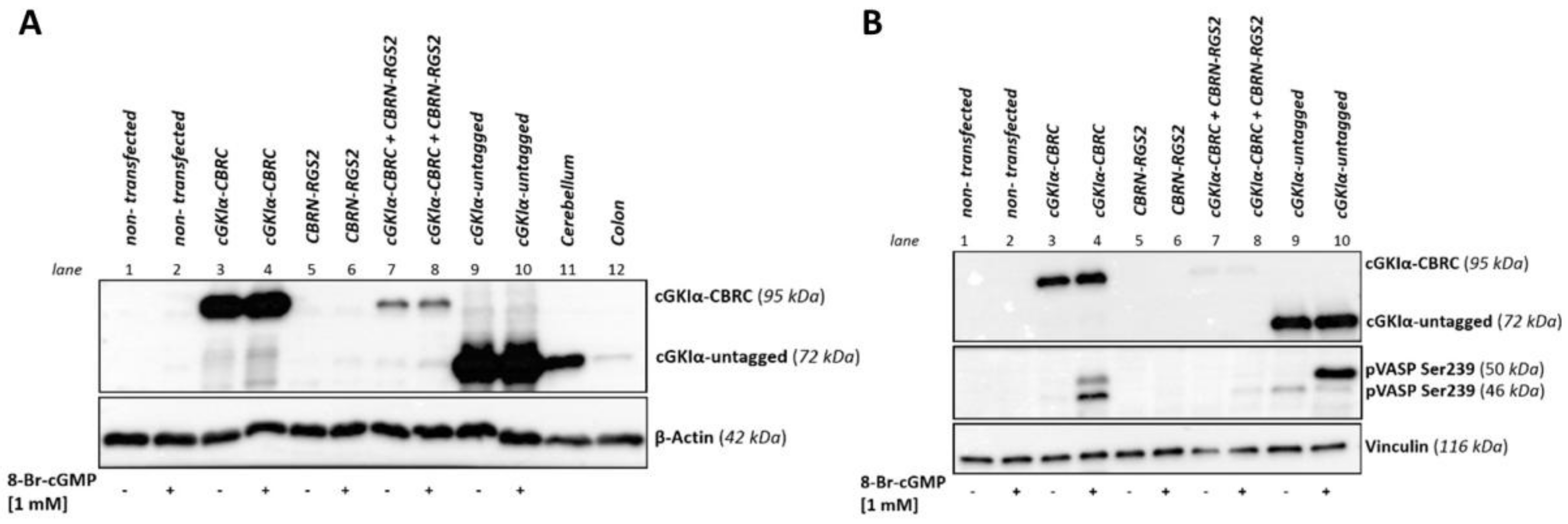
2.2. Expression in a Eukaryotic System
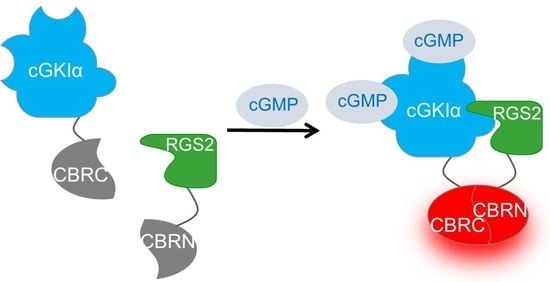
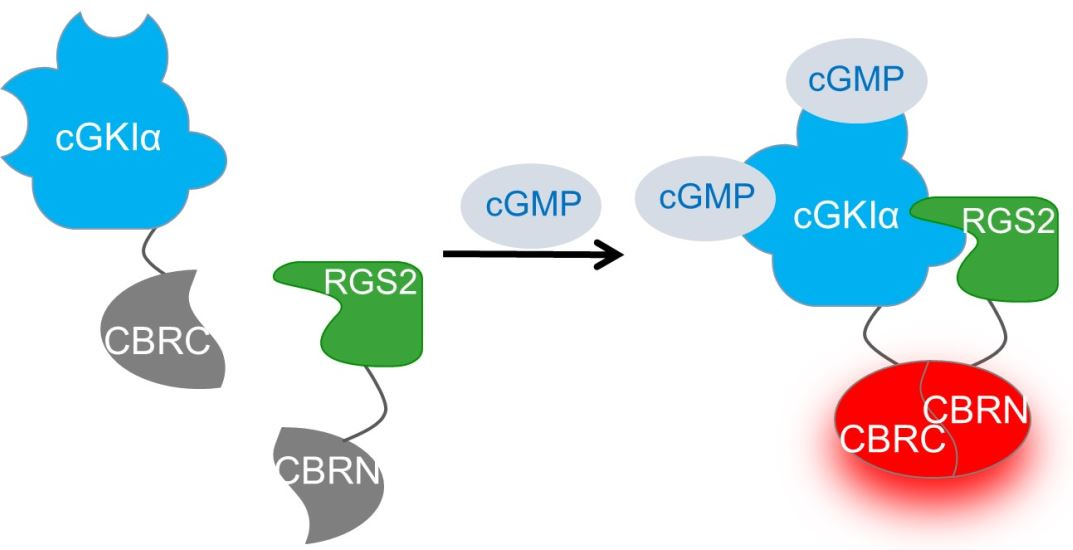
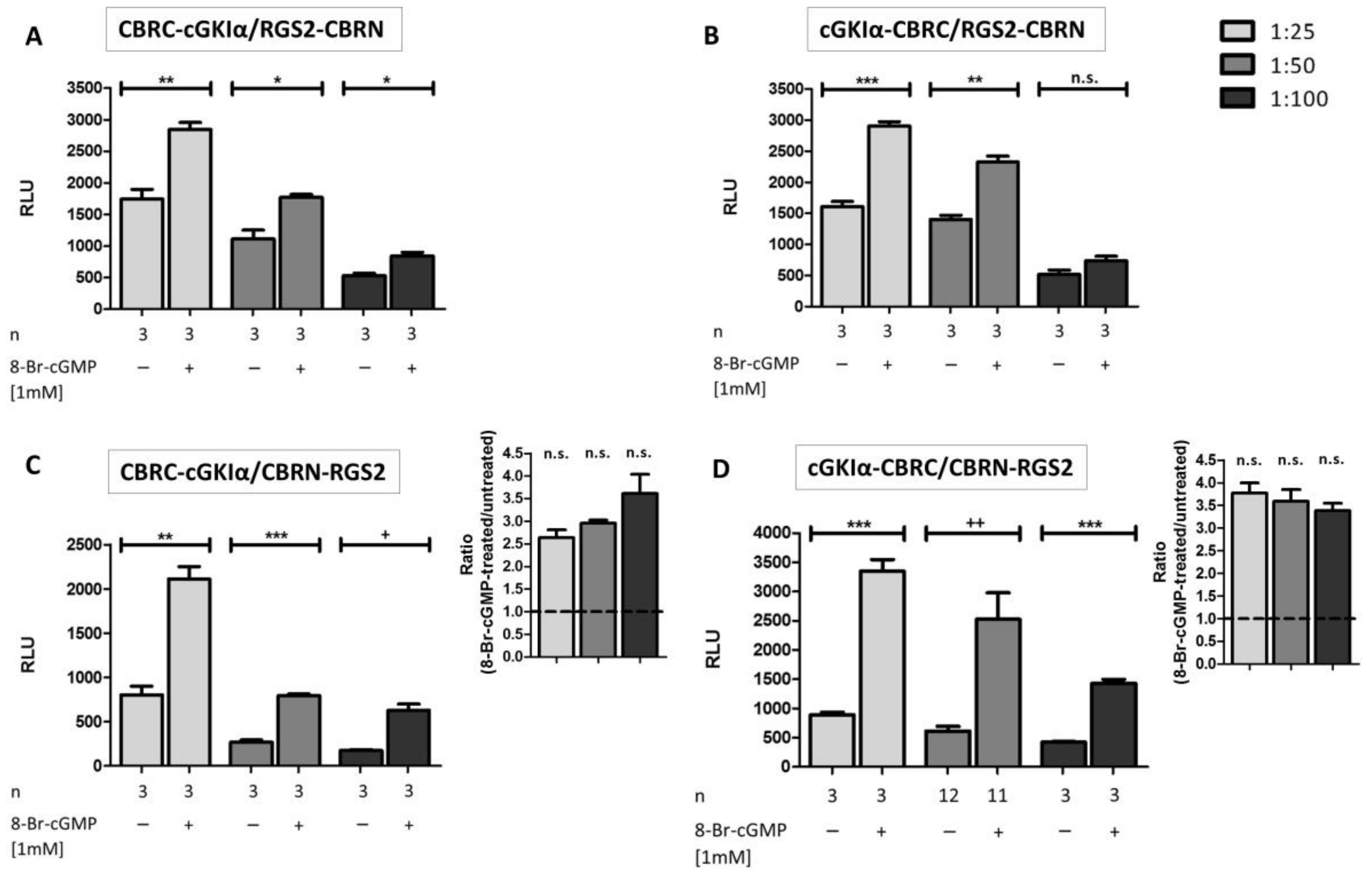
2.3. Interaction Analysis of cGKIα and RGS2
2.4. Comparison of Expression Level of cGKIα with Tissue Expression and Analysis of Activity
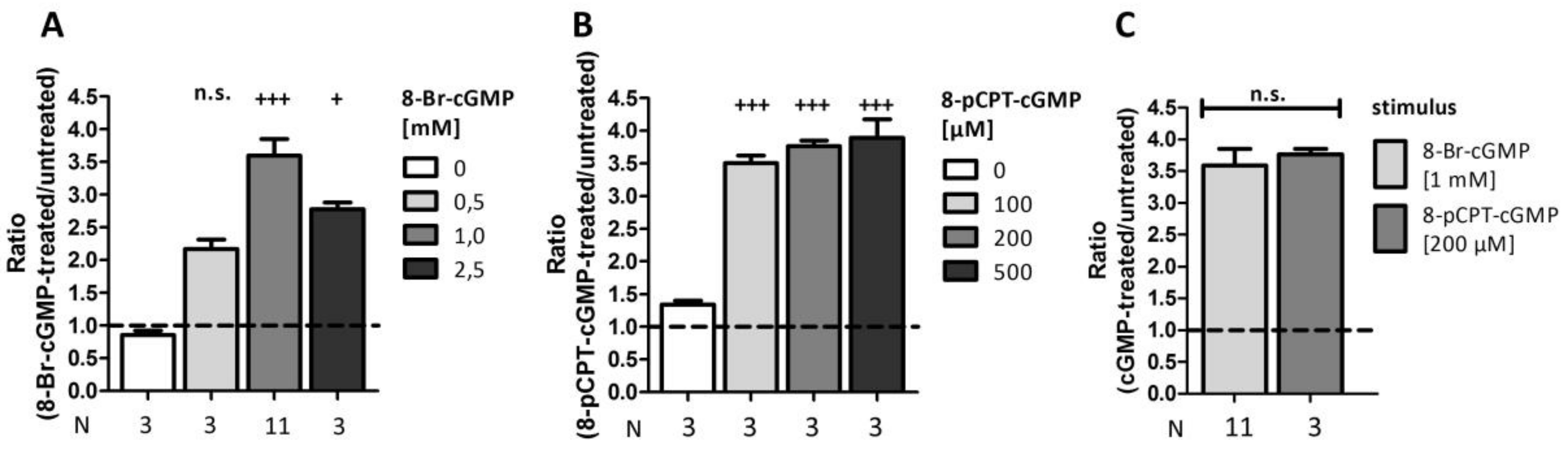
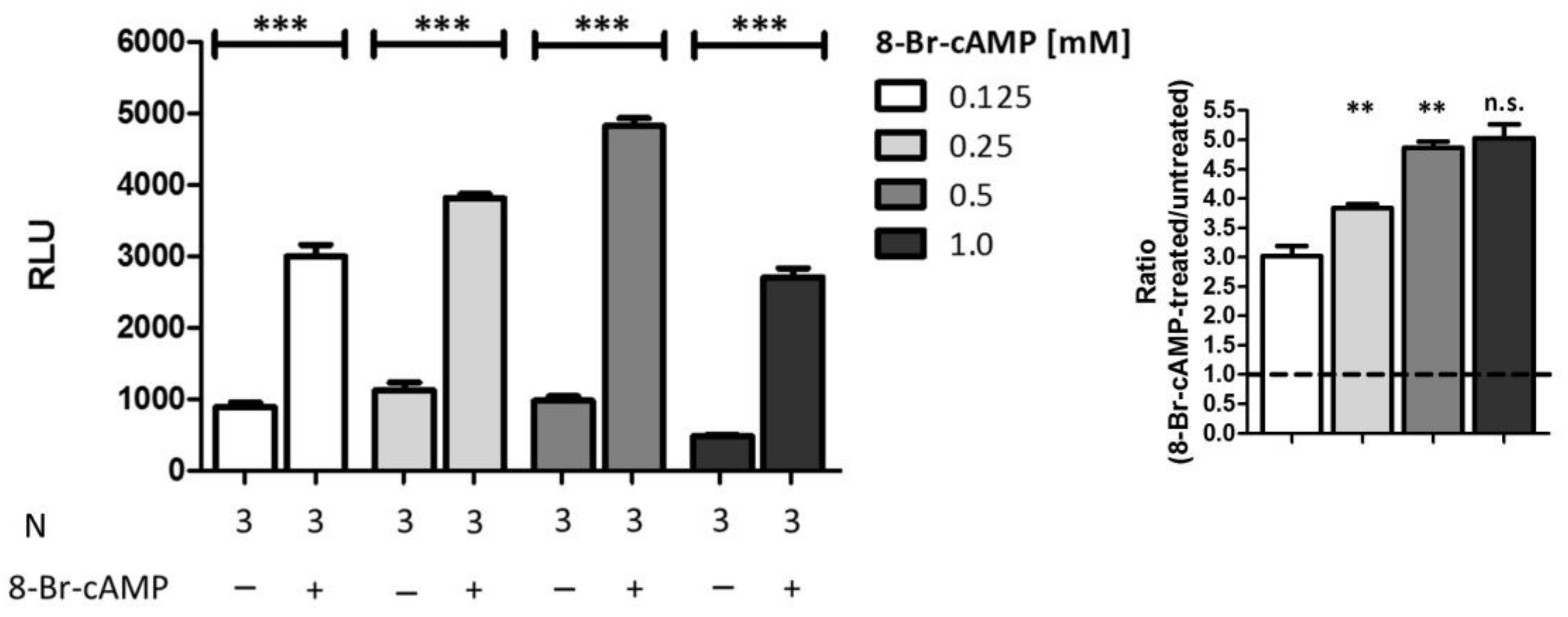
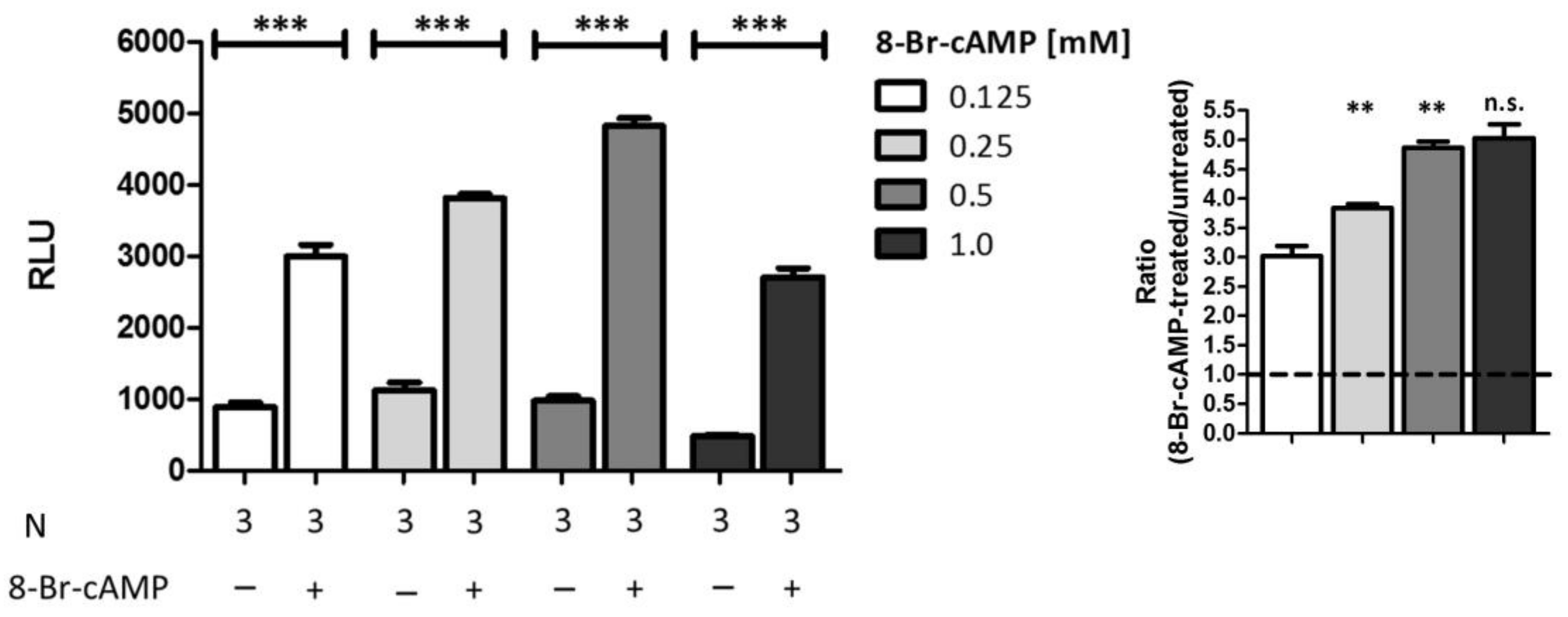
2.5. Comparison of Different Stimuli
2.6. Analysis of Specificity
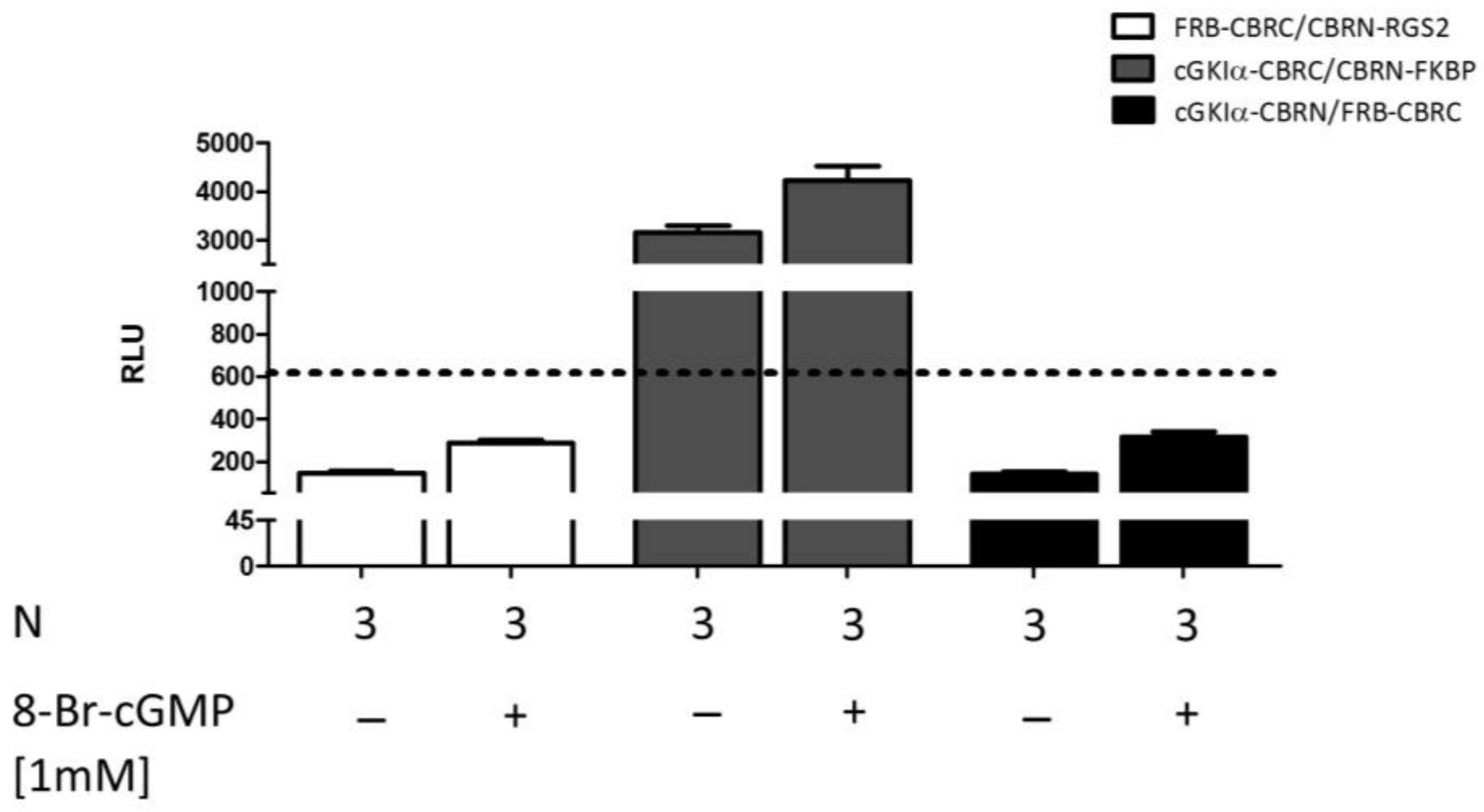
2.7. Analysis of Selectivity
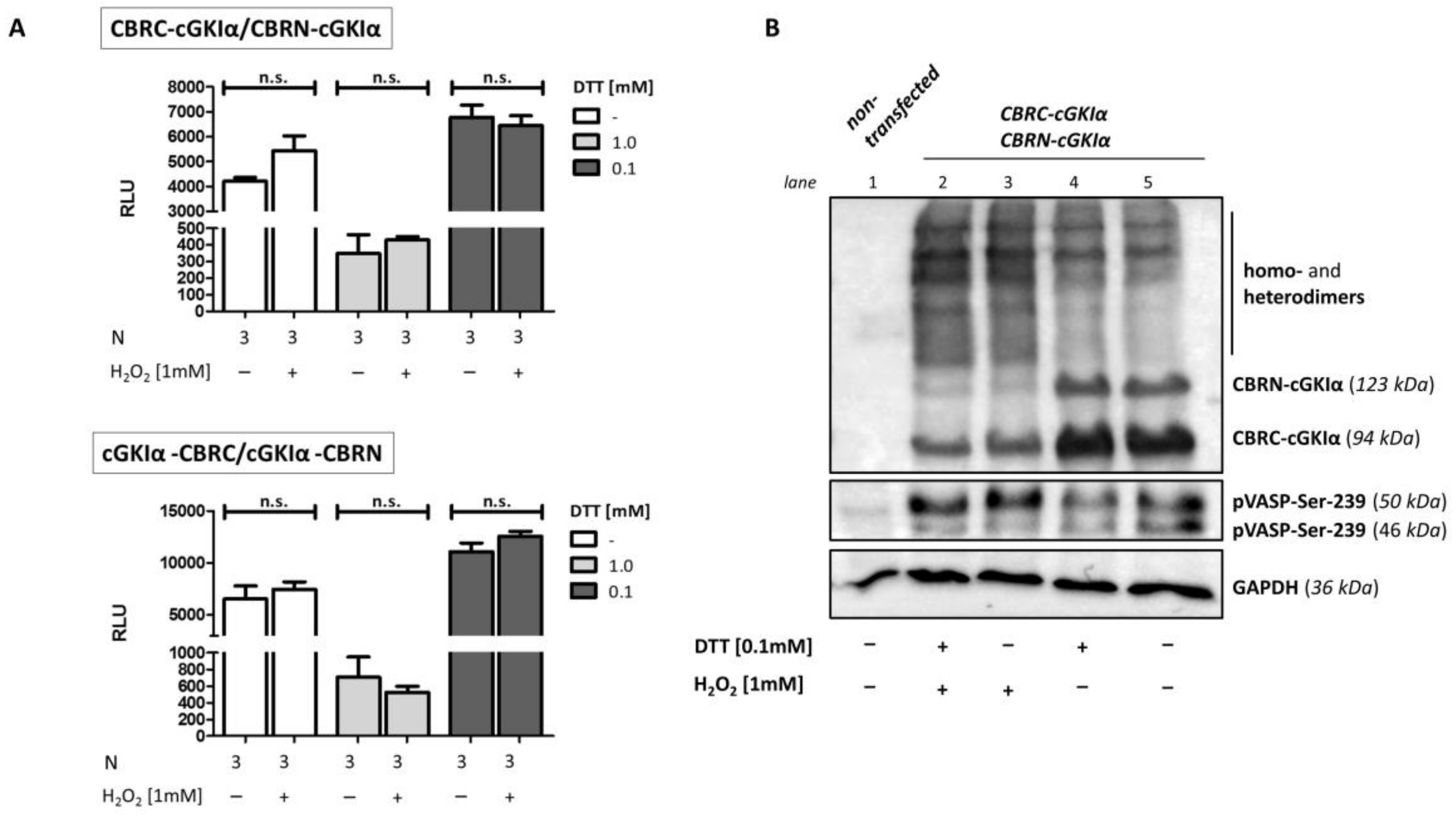
2.8. Dimerization of cGKIα upon Oxidant Stimulation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Construction of Vectors
4.3. Cell Culture and Transfection
4.4. Western Blotting
4.5. Luminescence Assay
4.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stumpf, M.P.H.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, N.; Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 10340–10344. [Google Scholar] [CrossRef] [PubMed]
- Stagljar, I.; Korostensky, C.; Johnsson, N.; te Heesen, S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 5187–5192. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Wilson, C.G.M.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.R.; Hillcoat, B.L. Regulation of dihydrofolate reductase and other folate-requiring enzymes. Adv. Enzyme Regul. 1968, 6, 335–349. [Google Scholar] [CrossRef]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein-protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Primeau, M.; Trudeau, L.-E.; Michnick, S.W. β-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 2002, 20, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Aquin, S.; Berger, N.; Landry, C.R.; Nyfeler, B.; Bouvier, M.; Michnick, S.W. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Michnick, S.W. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 2006, 3, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Smith, M.C.P.; Luker, G.D.; Gammon, S.T.; Piwnica-Worms, H.; Piwnica-Worms, D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S.S. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 2003, 75, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, V.; Naik, S.; Piwnica-Worms, D. Current state of imaging protein-protein interactions in vivo with genetically encoded reporters. Annu. Rev. Biomed. Eng. 2007, 9, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Viviani, V.R.; Arnoldi, F.G.C.; Neto, A.J.S.; Oehlmeyer, T.L.; Bechara, E.J.H.; Ohmiya, Y. The structural origin and biological function of pH-sensitivity in firefly luciferases. Photochem. Photobiol. Sci. 2008, 7, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Vaandrager, A.B.; Ehlert, E.M.; Jarchau, T.; Lohmann, S.M.; de Jonge, H.R. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J. Biol. Chem. 1996, 271, 7025–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernet, W.; Flockerzi, V.; Hofmann, F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989, 251, 191–196. [Google Scholar] [CrossRef]
- Orstavik, S.; Natarajan, V.; Taskén, K.; Jahnsen, T.; Sandberg, M. Characterization of the human gene encoding the type I alpha and type I beta cGMP-dependent protein kinase (PRKG1). Genomics 1997, 42, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Ruth, P.; Dostmann, W.; Sausbier, M.; Klatt, P.; Hofmann, F. Structure and function of cGMP-dependent protein kinases. Rev. Physiol. Biochem. Pharmacol. 1999, 135, 105–149. [Google Scholar] [PubMed]
- Hofmann, F.; Wegener, J.W. cGMP-dependent protein kinases (cGK). Methods Mol. Biol. 2013, 1020, 17–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lorenz, R.; Arold, S.T.; Reger, A.S.; Sankaran, B.; Casteel, D.E.; Herberg, F.W.; Kim, C. Crystal Structure of PKG I:cGMP Complex Reveals a cGMP-Mediated Dimeric Interface that Facilitates cGMP-Induced Activation. Structure 2016, 24, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Richie-Jannetta, R.; Busch, J.L.; Higgins, K.A.; Corbin, J.D.; Francis, S.H. Isolated regulatory domains of cGMP-dependent protein kinase Iα and Iβ retain dimerization and native cGMP-binding properties and undergo isoform-specific conformational changes. J. Biol. Chem. 2006, 281, 6977–6984. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, W.; Regulla, S.; Meyer, H.E.; Hofmann, F. Oxidation of cysteines activates cGMP-dependent protein kinase. J. Biol. Chem. 1991, 266, 16305–16311. [Google Scholar] [PubMed]
- Zhang, D.X.; Borbouse, L.; Gebremedhin, D.; Mendoza, S.A.; Zinkevich, N.S.; Li, R.; Gutterman, D.D. H2O2-induced dilation in human coronary arterioles: Role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ. Res. 2012, 110, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, H.; Zhuang, S.; Pilz, R.B.; Casteel, D.E. The activity of cGMP-dependent protein kinase Iα is not directly regulated by oxidation-induced disulfide formation at cysteine 43. J. Biol. Chem. 2017, 292, 8262–8268. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.M.; Wang, G.; Lu, P.; Karas, R.H.; Aronovitz, M.; Heximer, S.P.; Kaltenbronn, K.M.; Blumer, K.J.; Siderovski, D.P.; Zhu, Y.; et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 2003, 9, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, H.G.; Apaniesk, D.; Chen, Y.; Song, J.; Nusskern, D. Inhibition of G-protein signaling by dominant gain-of-function mutations in Sst2p, a pheromone desensitization factor in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995, 15, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Heximer, S.P.; Knutsen, R.H.; Sun, X.; Kaltenbronn, K.M.; Rhee, M.-H.; Peng, N.; Oliveira-dos-Santos, A.; Penninger, J.M.; Muslin, A.J.; Steinberg, T.H.; et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Investig. 2003, 111, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Osei-Owusu, P.; Sun, X.; Drenan, R.M.; Steinberg, T.H.; Blumer, K.J. Regulation of RGS2 and Second Messenger Signaling in Vascular Smooth Muscle Cells by cGMP-dependent Protein Kinase. J. Biol. Chem. 2007, 282, 31656–31665. [Google Scholar] [CrossRef] [PubMed]
- Hida, N.; Awais, M.; Takeuchi, M.; Ueno, N.; Tashiro, M.; Takagi, C.; Singh, T.; Hayashi, M.; Ohmiya, Y.; Ozawa, T. High-sensitivity real-time imaging of dual protein-protein interactions in living subjects using multicolor luciferases. PLoS ONE 2009, 4, e5868. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N.; Kafi, A.K.M.; Hattori, M.; Miura, K.; Masuda, K.; Ozawa, T. Rapid and high-sensitivity cell-based assays of protein-protein interactions using split click beetle luciferase complementation: An approach to the study of G-protein-coupled receptors. Anal. Chem. 2010, 82, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, V.; Naik, S.; Bruinsma, M.; Dothager, R.S.; Pan, M.-H.; Samrakandi, M.; Moss, B.; Elhammali, A.; Piwnica-Worms, D. Dual-color click beetle luciferase heteroprotein fragment complementation assays. Chem. Biol. 2010, 17, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Anton, A.; Salim, S.; Blumer, K.J.; Dessauer, C.W.; Heximer, S.P. Alternative translation initiation of human regulators of G-protein signaling-2 yields a set of functionally distinct proteins. Mol. Pharmacol. 2008, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Mueller-Thuemen, P.; Littmann, T.; Harloff, M.; Ozawa, T.; Schlossmann, J. University of Regensburg, Regensburg, Germany, Unpublished work, 2016.
- Halbrügge, M.; Friedrich, C.; Eigenthaler, M.; Schanzenbächer, P.; Walter, U. Stoichiometric and reversible phosphorylation of a 46-kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J. Biol. Chem. 1990, 265, 3088–3093. [Google Scholar] [PubMed]
- Butt, E.; Abel, K.; Krieger, M.; Palm, D.; Hoppe, V.; Hoppe, J.; Walter, U. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 1994, 269, 14509–14517. [Google Scholar] [PubMed]
- Richie-Jannetta, R.; Francis, S.H.; Corbin, J.D. Dimerization of cGMP-dependent protein kinase Iβ is mediated by an extensive amino-terminal leucine zipper motif, and dimerization modulates enzyme function. J. Biol. Chem. 2003, 278, 50070–50079. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-Y.; Cohen, E.D.; Genieser, H.-G.; Barnstable, C.J. Substituted cGMP analogs can act as selective agonists of the rod photoreceptor cGMP-gated cation channel. J. Mol. Neurosci. 1998, 10, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Donahoe, P.K.; Zervos, A.S. Specific interaction of type I receptors of the TGF-β family with the immunophilin FKBP-12. Science 1994, 265, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Hardman, J.G. Cyclic nucleotides and regulation of vascular smooth muscle. J. Cardiovasc. Pharmacol. 1984, 6 (Suppl. 4), S639–S645. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Noblett, B.D.; Todd, B.W.; Wells, J.N.; Corbin, J.D. Relaxation of vascular and tracheal smooth muscle by cyclic nucleotide analogs that preferentially activate purified cGMP-dependent protein kinase. Mol. Pharmacol. 1988, 34, 506–517. [Google Scholar] [PubMed]
- Lincoln, T.M.; Cornwell, T.L.; Taylor, A.E. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. Am. J. Physiol. 1990, 258, C399–C407. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Kryman, J.P.; El-Mowafy, A.M.; Han, G.; Carrier, G.O. cAMP-dependent vasodilators cross-activate the cGMP-dependent protein kinase to stimulate BK(Ca) channel activity in coronary artery smooth muscle cells. Circ. Res. 2000, 86, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Shabb, J.B.; Ng, L.; Corbin, J.D. One amino acid change produces a high affinity cGMP-binding site in cAMP-dependent protein kinase. J. Biol. Chem. 1990, 265, 16031–16034. [Google Scholar] [PubMed]
- Kim, J.J.; Casteel, D.E.; Huang, G.; Kwon, T.H.; Ren, R.K.; Zwart, P.; Headd, J.J.; Brown, N.G.; Chow, D.-C.; Palzkill, T.; et al. Co-crystal structures of PKG Iβ (92-227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PLoS ONE 2011, 6, e18413. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.Y.; Kim, J.J.; Reger, A.S.; Lorenz, R.; Moon, E.-W.; Zhao, C.; Casteel, D.E.; Bertinetti, D.; Vanschouwen, B.; Selvaratnam, R.; et al. Structural basis for cyclic-nucleotide selectivity and cGMP-selective activation of PKG I. Structure 2014, 22, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, B.; Kramer, R.H. Real-time patch-cram detection of intracellular cGMP reveals long-term suppression of responses to NO and muscarinic agonists. Neuron 1998, 21, 895–906. [Google Scholar] [CrossRef]
- Iancu, R.V.; Ramamurthy, G.; Warrier, S.; Nikolaev, V.O.; Lohse, M.J.; Jones, S.W.; Harvey, R.D. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am. J. Physiol. Cell Physiol. 2008, 295, C414–C422. [Google Scholar] [CrossRef] [PubMed]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.; Okumura, K.; Omori, K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Prysyazhna, O.; Rudyk, O.; Eaton, P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 2012, 18, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Scotcher, J.; Prysyazhna, O.; Boguslavskyi, A.; Kistamas, K.; Hadgraft, N.; Martin, E.D.; Worthington, J.; Rudyk, O.; Rodriguez Cutillas, P.; Cuello, F.; et al. Disulfide-activated protein kinase G Iα regulates cardiac diastolic relaxation and fine-tunes the Frank-Starling response. Nat. Commun. 2016, 7, 13187. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; DePinho, R.A. Cellular senescence: mitotic clock or culture shock? Cell 2000, 102, 407–410. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett. 2003, 540, 3–6. [Google Scholar] [CrossRef]
- Hua Long, L.; Halliwell, B. Oxidation and generation of hydrogen peroxide by thiol compounds in commonly used cell culture media. Biochem. Biophys. Res. Commun. 2001, 286, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Müller, P.M.; Gnügge, R.; Dhayade, S.; Thunemann, M.; Krippeit-Drews, P.; Drews, G.; Feil, R. H2O2 lowers the cytosolic Ca2+ concentration via activation of cGMP-dependent protein kinase Iα. Free Radic. Biol. Med. 2012, 53, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ranek, M.J.; Lee, D.I.; Shalkey Hahn, V.; Kim, C.; Eaton, P.; Kass, D.A. Prevention of PKG1α oxidation augments cardioprotection in the stressed heart. J. Clin. Investig. 2015, 125, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Ruth, P.; Pfeifer, A.; Kamm, S.; Klatt, P.; Dostmann, W.R.; Hofmann, F. Identification of the amino acid sequences responsible for high affinity activation of cGMP kinase Iα. J. Biol. Chem. 1997, 272, 10522–10528. [Google Scholar] [CrossRef] [PubMed]
- Geiselhöringer, A.; Gaisa, M.; Hofmann, F.; Schlossmann, J. Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS Lett. 2004, 575, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; van der Eb, A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schramm, A.; Mueller-Thuemen, P.; Littmann, T.; Harloff, M.; Ozawa, T.; Schlossmann, J. Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies. Int. J. Mol. Sci. 2018, 19, 1180. https://doi.org/10.3390/ijms19041180
Schramm A, Mueller-Thuemen P, Littmann T, Harloff M, Ozawa T, Schlossmann J. Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies. International Journal of Molecular Sciences. 2018; 19(4):1180. https://doi.org/10.3390/ijms19041180
Chicago/Turabian StyleSchramm, Andrea, Philip Mueller-Thuemen, Timo Littmann, Manuela Harloff, Takeaki Ozawa, and Jens Schlossmann. 2018. "Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies" International Journal of Molecular Sciences 19, no. 4: 1180. https://doi.org/10.3390/ijms19041180
APA StyleSchramm, A., Mueller-Thuemen, P., Littmann, T., Harloff, M., Ozawa, T., & Schlossmann, J. (2018). Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies. International Journal of Molecular Sciences, 19(4), 1180. https://doi.org/10.3390/ijms19041180