Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches


Abstract
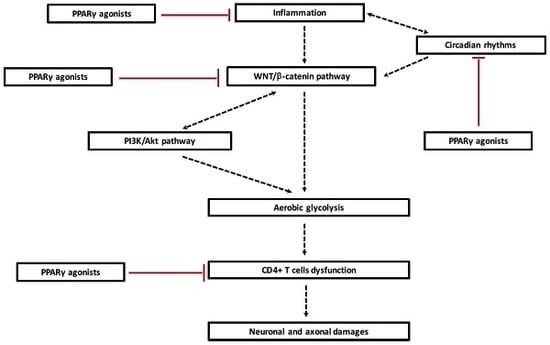
:
1. Introduction
2. PPARγ
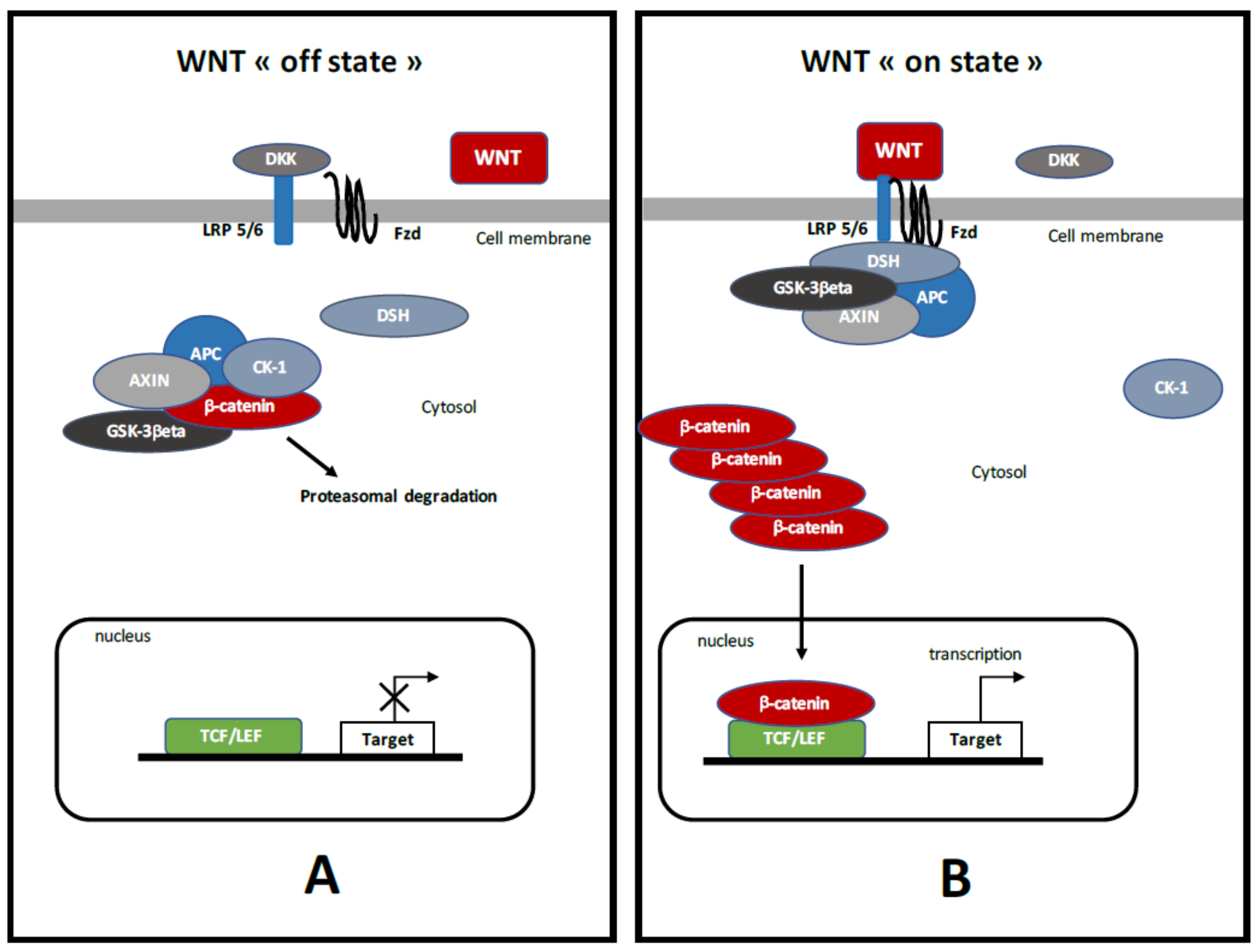
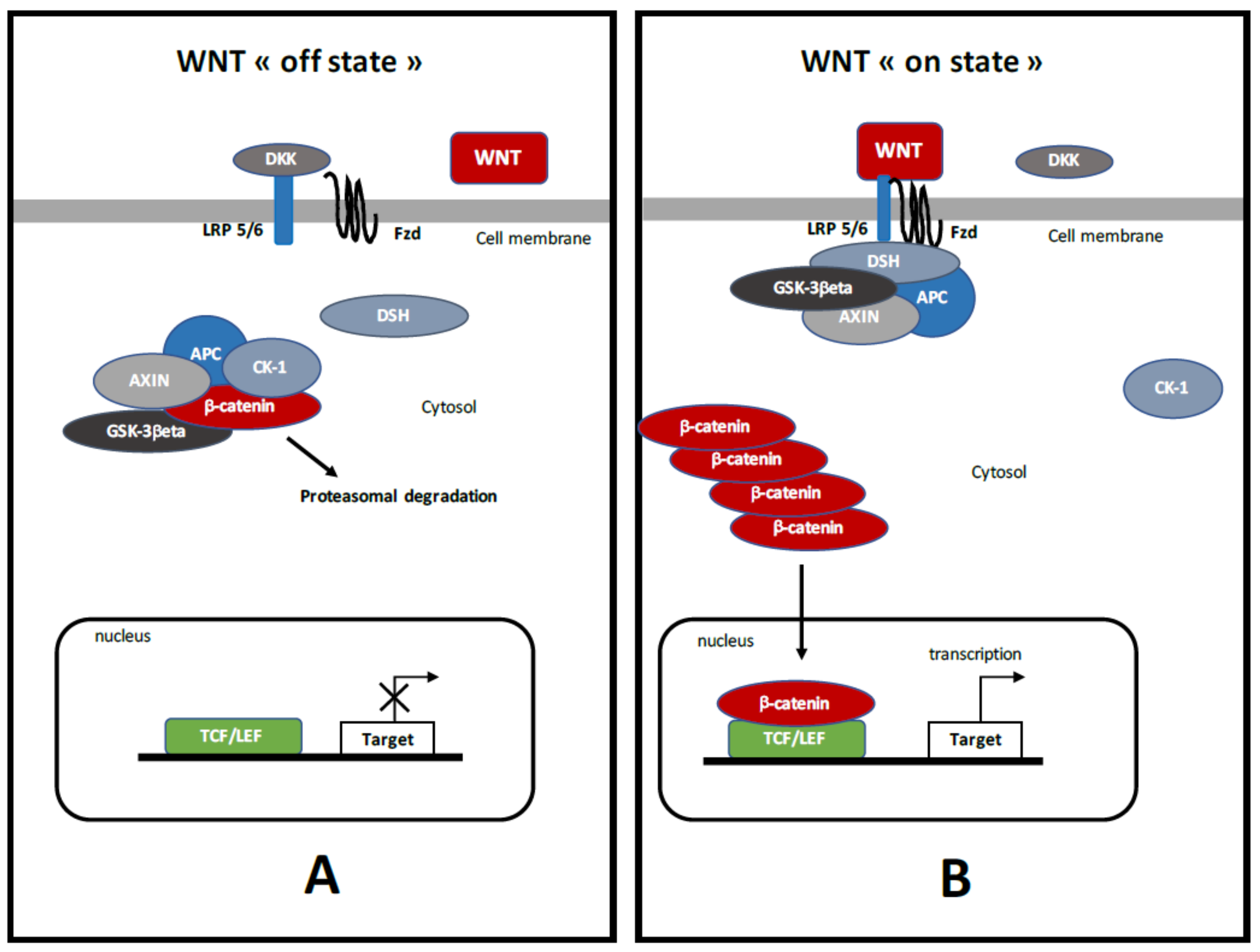
3. Canonical WNT/β-Catenin Pathway (Figure 1)
4. Crosstalk between PPARγ and Canonical WNT/β-Catenin Pathway in Diseases
5. PPARγ and the Canonical WNT/β-Catenin Pathway in MS
5.1. PPARγ in MS
5.2. Demyelination and Activation of WNT/β-Catenin Pathway
5.3. Opposed Interaction between PPARγ and WNT Pathway in MS
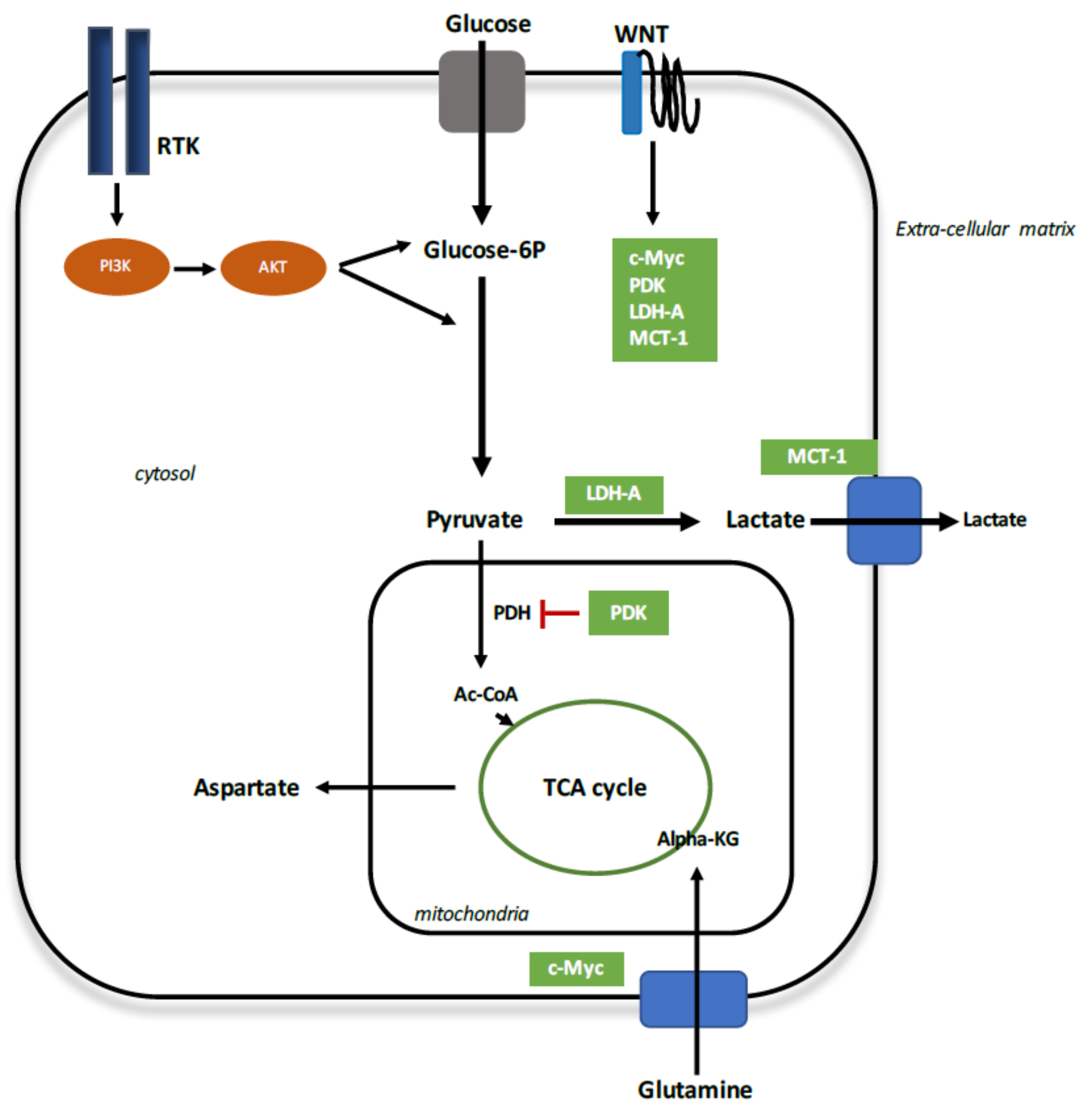
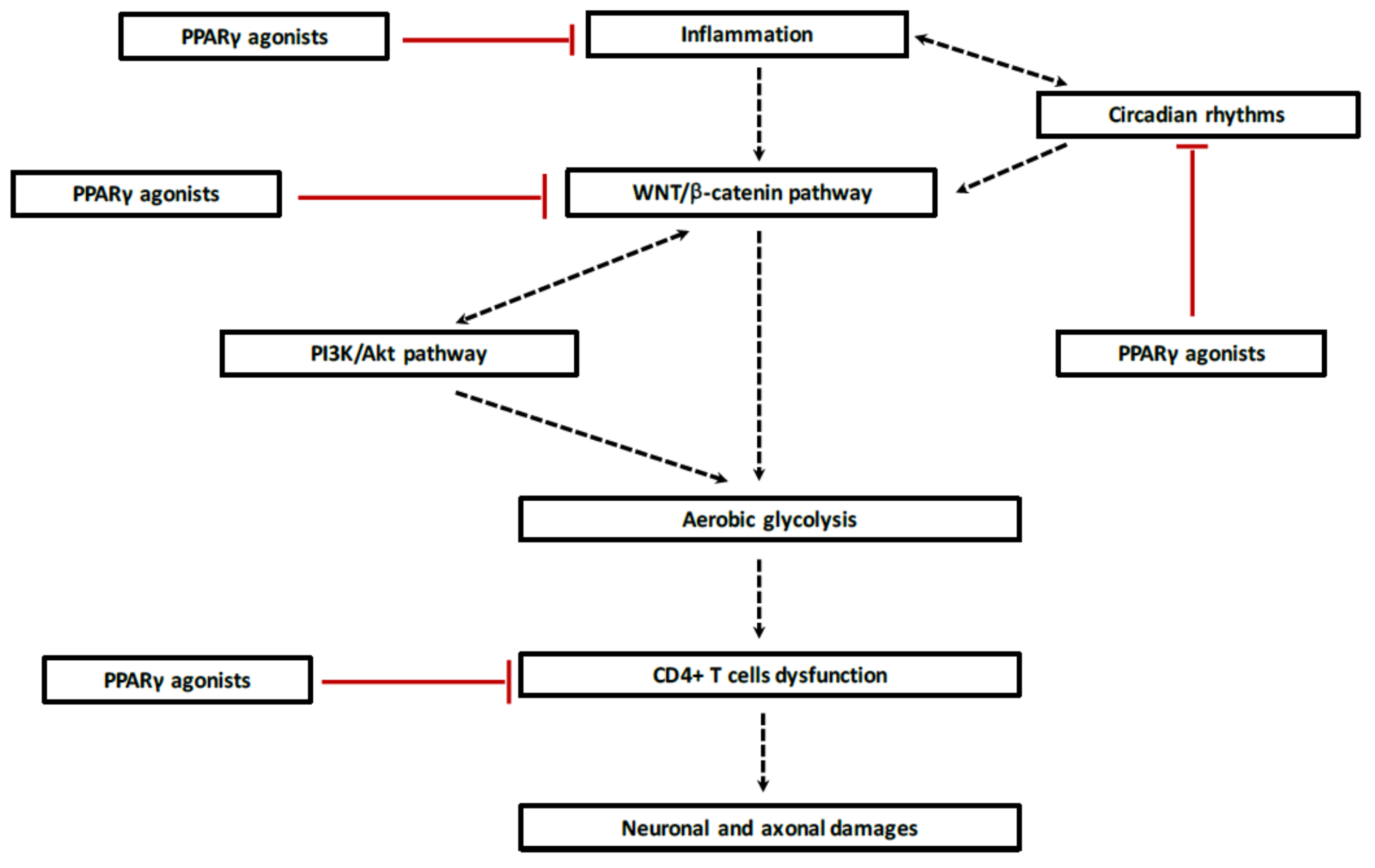
6. Reprogramming Energy Metabolism in Demyelination
6.1. Aerobic Glycolysis
6.2. Aerobic Glycolysis in MS
7. Circadian Rhythms in MS
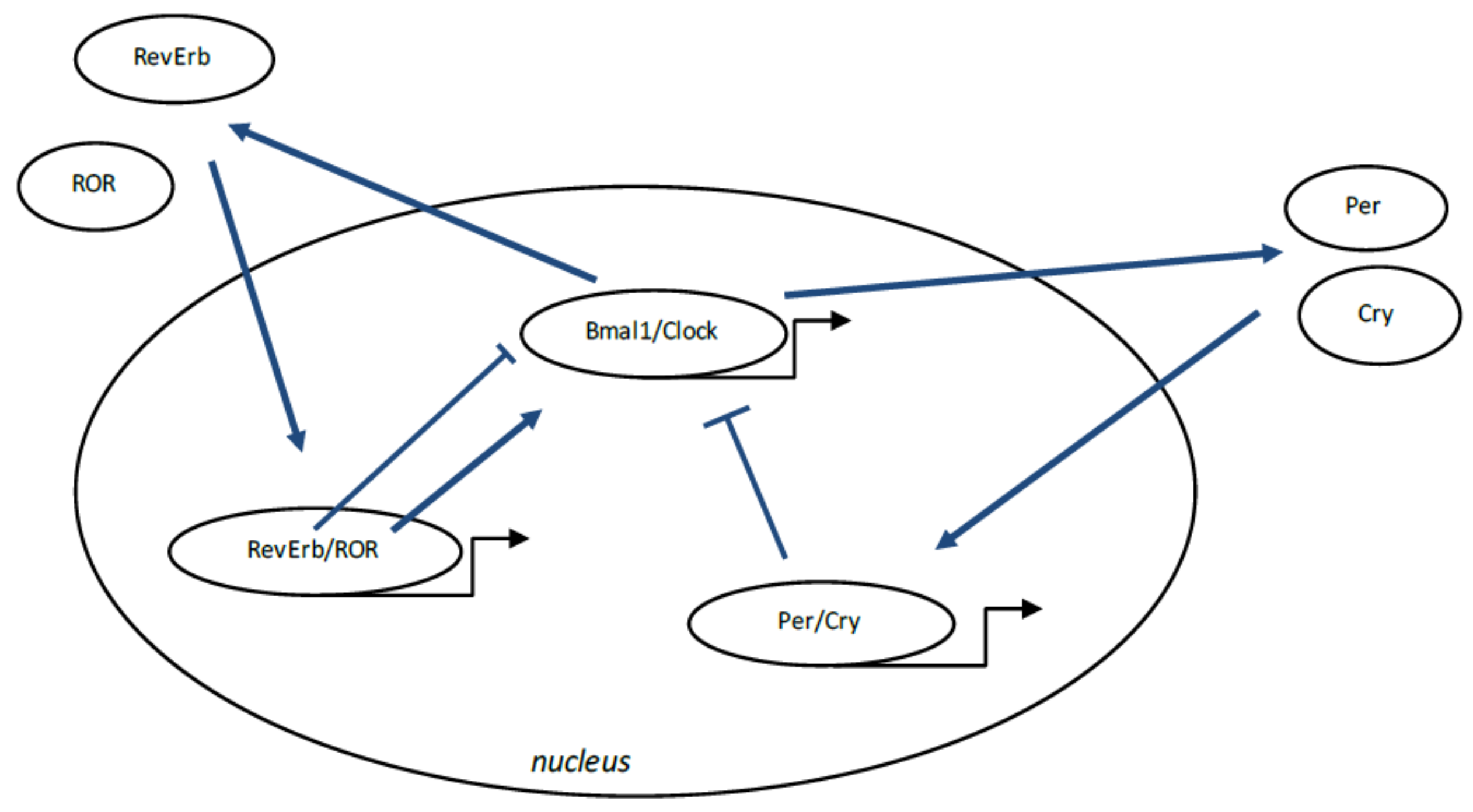
7.1. Circadian Rhythms, Definition
7.2. Circadian Rhythm Disruption in MS
7.3. Interaction between WNT/β-Catenin Pathway and Circadian Rhythms
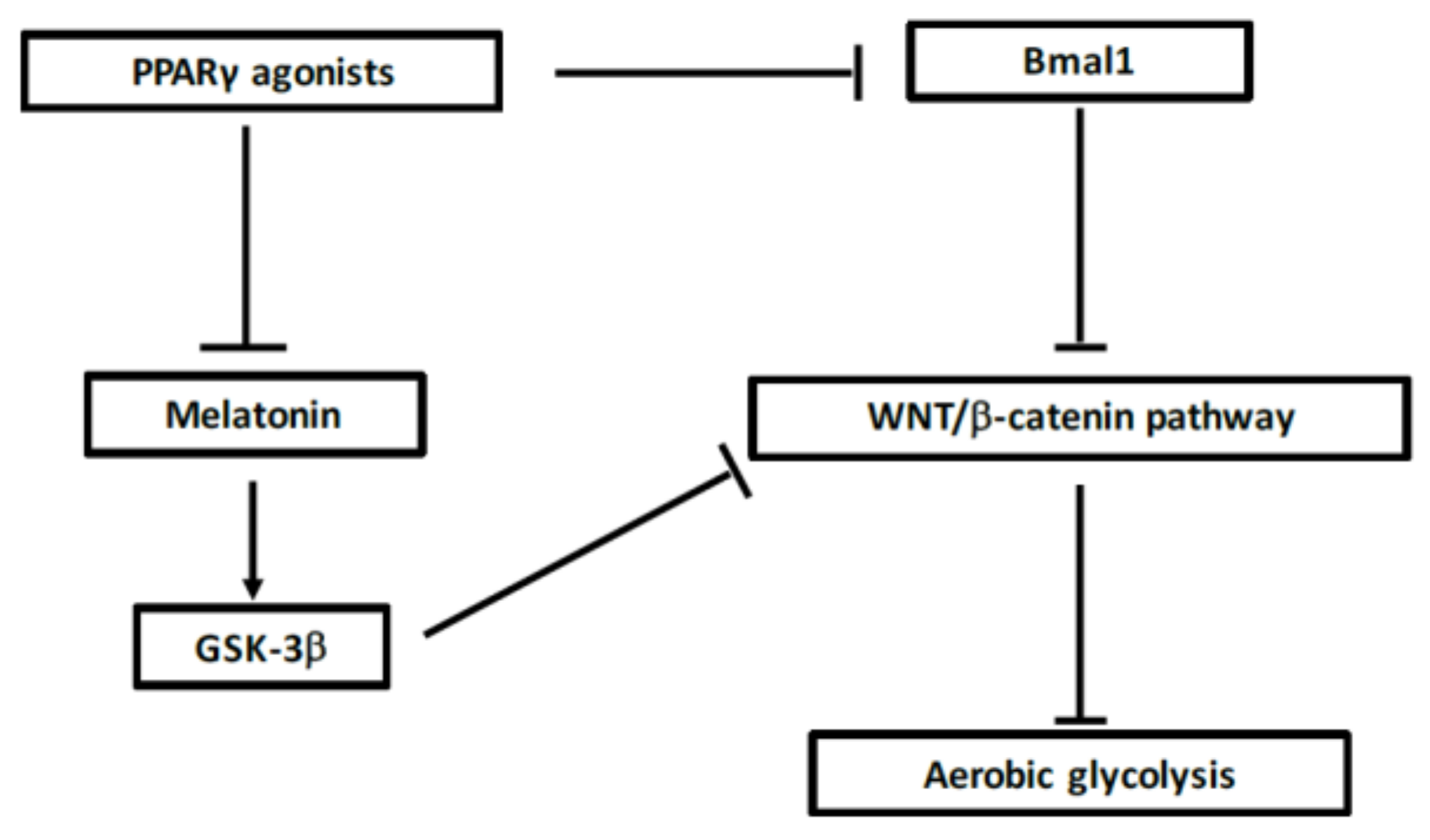
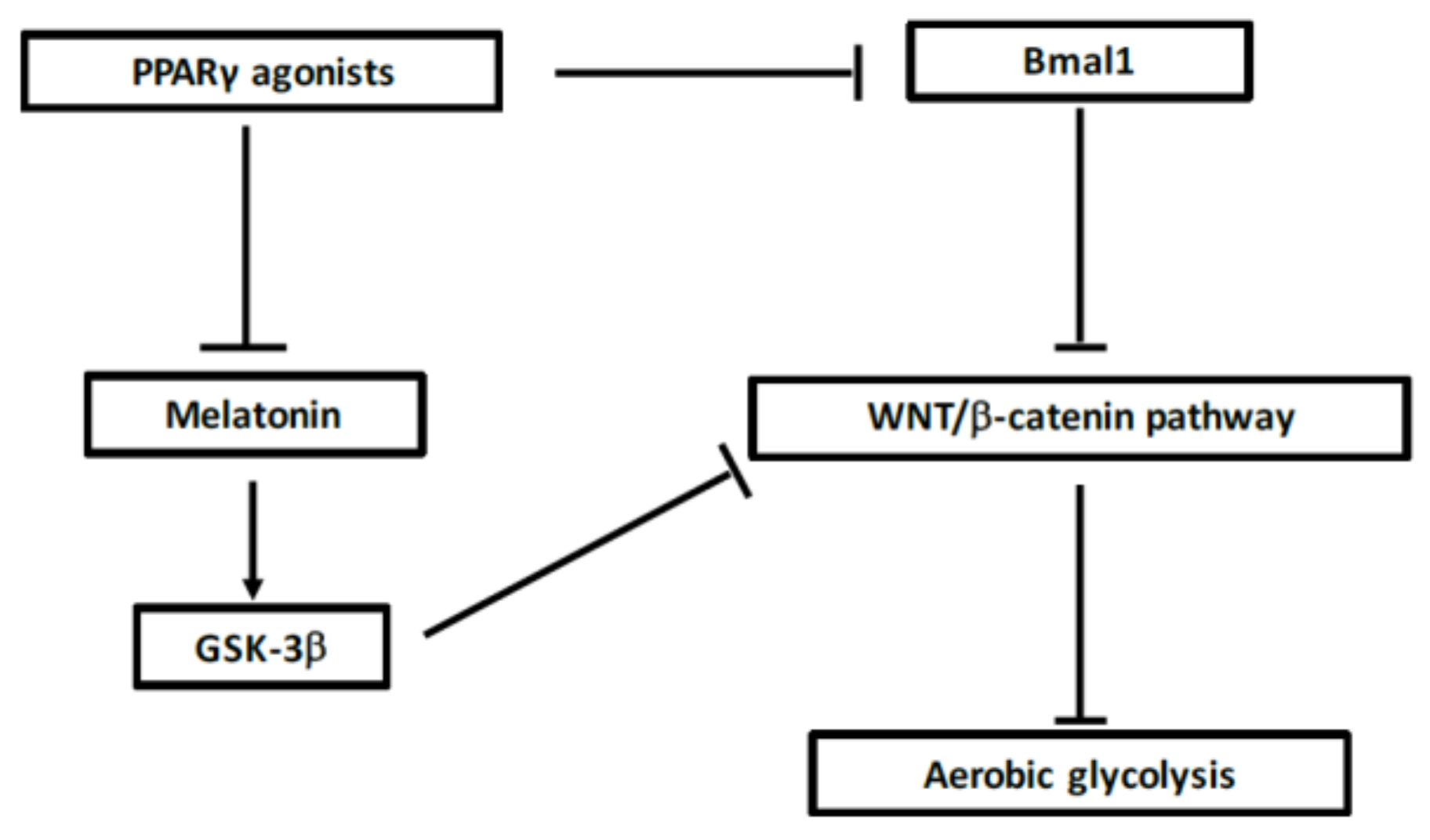
7.4. Action of PPARγ on Circadian Rhythms
7.5. Interest of Cortisol in MS
7.6. Interest of Melatonin in MS
8. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| Acetyl-coA | Acetyl-coenzyme A |
| APC | Adenomatous polyposis coli |
| Bmal1 | Brain and muscle aryl-hydrocarbon receptor nuclear translocator-like 1 |
| Clock | Circadian locomotor output cycles kaput |
| Cry | Cryptochrome |
| CRs | Circadian rhythms |
| DSH | Disheveled |
| FZD | Frizzled |
| Glut | Glucose transporter |
| GSK-3β | Glycogen synthase kinase-3β |
| LDH | Lactate dehydrogenase |
| LRP 5/6 | Low-density lipoprotein receptor-related protein 5/6 |
| MCT-1 | Monocarboxylate lactate transporter-1 |
| Per | Period |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PI3K-Akt | Phosphatidylinositol 3-kinase-protein kinase B |
| PDH | Pyruvate dehydrogenase complex |
| PDK | Pyruvate dehydrogenase kinase |
| RORs | Retinoid-related orphan receptors |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TCA | Tricarboxylic acid |
References
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.-A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.; Wang, Y.; Kivisäkk, P.; Bronson, R.T.; Meyer, M.; Imitola, J.; Khoury, S.J. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing—Remitting experimental autoimmune encephalomyelitis. Brain J. Neurol. 2007, 130, 2816–2829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Oksenberg, J.R. Genetic determinants of risk and progression in multiple sclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 449, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef] [PubMed]
- Woodward, L.J.; Anderson, P.J.; Austin, N.C.; Howard, K.; Inder, T.E. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 2006, 355, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Rosenberg, P.A. Pathophysiology of glia in perinatal white matter injury. Glia 2014, 62, 1790–1815. [Google Scholar] [CrossRef] [PubMed]
- Billiards, S.S.; Haynes, R.L.; Folkerth, R.D.; Borenstein, N.S.; Trachtenberg, F.L.; Rowitch, D.H.; Ligon, K.L.; Volpe, J.J.; Kinney, H.C. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 2008, 18, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.J.; Kotter, M.R.; Harrington, E.P.; Huang, J.K.; Zhao, C.; Rowitch, D.H.; Franklin, R.J.M. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp. Neurol. 2010, 225, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain J. Neurol. 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Sandler, S. Chemical and Engineering Thermodynamics, 4th ed.; Wiely: New York, NY, USA, 2006. [Google Scholar]
- Garcia, H.G.; Kondev, J.; Orme, N.; Theriot, J.A.; Phillips, R. Thermodynamics of biological processes. Methods Enzymol. 2011, 492, 27–59. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N.; Guillevin, R.; Lecarpentier, Y. Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma on Neuroinflammation, Demyelination, and Remyelination in Multiple Sclerosis. Cell. Mol. Neurobiol. 2017, 38, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Tsentidis, C.; Gourgiotis, D.; Kossiva, L.; Marmarinos, A.; Doulgeraki, A.; Karavanaki, K. Increased levels of Dickkopf-1 are indicative of Wnt/β-catenin downregulation and lower osteoblast signaling in children and adolescents with type 1 diabetes mellitus, contributing to lower bone mineral density. Osteoporos. Int. 2017, 28, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Pörksen, S.; Nielsen, L.B.; Mortensen, H.B.; Danne, T.; Kocova, M.; Castaño, L.; Pociot, F.; Hougaard, P.; Ekstrøm, C.T.; Gammeltoft, S.; et al. Hvidøre Study Group on Childhood Diabetes Variation within the PPARG gene is associated with residual beta-cell function and glycemic control in children and adolescents during the first year of clinical type 1 diabetes. Pediatr. Diabetes 2008, 9, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Fallahi, P.; Vita, R.; Antonelli, A.; Benvenga, S. Peroxisome Proliferator-Activated Receptor-γ in Thyroid Autoimmunity. PPAR Res. 2015, 2015, 232818. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Ayala-Haedo, J.A.; Field, M.G.; Pelaez, D.; Wester, S.T. RNA-Sequencing Gene Expression Profiling of Orbital Adipose-Derived Stem Cell Population Implicate HOX Genes and WNT Signaling Dysregulation in the Pathogenesis of Thyroid-Associated Orbitopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6146–6158. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.Y.; Pan, Y.F.; Guo, X.H.; Wu, Y.Q.; Gu, J.R.; Cai, D.Z. Expression of β-catenin in rheumatoid arthritis fibroblast-like synoviocytes. Scand. J. Rheumatol. 2011, 40, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Marder, W.; Khalatbari, S.; Myles, J.D.; Hench, R.; Lustig, S.; Yalavarthi, S.; Parameswaran, A.; Brook, R.D.; Kaplan, M.J. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J. Am. Heart Assoc. 2013, 2, e000441. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Krokidis, X.; Martin, P.; Pineau, T.; Hébert, J.-L.; Quillard, J.; Cortes-Morichetti, M.; Coirault, C. Increased entropy production in diaphragm muscle of PPAR alpha knockout mice. J. Theor. Biol. 2008, 250, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Rone, M.B.; Cui, Q.-L.; Fang, J.; Wang, L.-C.; Zhang, J.; Khan, D.; Bedard, M.; Almazan, G.; Ludwin, S.K.; Jones, R.; et al. Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4698–4707. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Wnt meets Warburg: Another piece in the puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, M.-T.; Tang, Y.; Chen, Y.; Jiang, H.; Jones, T.T.; Rao, K.; Brewer, G.J.; Singh, K.K.; Nie, D. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L). Cancer Biol. Ther. 2010, 9, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Newland, P.; Starkweather, A.; Sorenson, M. Central fatigue in multiple sclerosis: A review of the literature. J. Spinal Cord Med. 2016, 39, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yang, G.; Jia, Z.; Zhang, H.; Aoyagi, T.; Soodvilai, S.; Symons, J.D.; Schnermann, J.B.; Gonzalez, F.J.; Litwin, S.E.; et al. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab. 2008, 8, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromolecular Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R.J. A role for central nervous system PPAR-γ in the regulation of energy balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Blandon, J.; Rude, J.; Elfar, A.; Mukherjee, D. PPAR-γ agonist in treatment of diabetes: Cardiovascular safety considerations. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Oyekan, A. PPARs and their effects on the cardiovascular system. Clin. Exp. Hypertens. 2011, 33, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Montague, C.T.; Prins, J.B.; Holder, J.C.; Smith, S.A.; Sanders, L.; Digby, J.E.; Sewter, C.P.; Lazar, M.A.; Chatterjee, V.K.; et al. Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J. Clin. Investig. 1997, 100, 3149–3153. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Takano, H.; Nagai, T.; Uozumi, H.; Hasegawa, H.; Kubota, N.; Saito, T.; Masuda, Y.; Kadowaki, T.; Komuro, I. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation 2002, 105, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Miriyala, S.; Panchatcharam, M. PPARγ and Its Role in Cardiovascular Diseases. PPAR Res. 2017, 2017, 6404638. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liang, F.; Moriya, J.; Yamakawa, J.; Takahashi, T.; Shen, L.; Kanda, T. Peroxisome proliferator-activated receptors (PPARs) and their agonists for hypertension and heart failure: Are the reagents beneficial or harmful? Int. J. Cardiol. 2008, 130, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Galli, A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res. 2012, 2012, 641087. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Minghetti, L. Regulation of Glial Cell Functions by PPAR-gamma Natural and Synthetic Agonists. PPAR Res. 2008, 2008, 864140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lovett-Racke, A.E.; Racke, M.K. Regulation of Immune Responses and Autoimmune Encephalomyelitis by PPARs. PPAR Res. 2010, 2010, 104705. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Diehl, L.; Dani, I.; Neumann, H.; von Oppen, N.; Dolf, A.; Endl, E.; Klockgether, T.; Engelhardt, B.; Knolle, P. Brain endothelial PPARgamma controls inflammation-induced CD4+ T cell adhesion and transmigration in vitro. J. Neuroimmunol. 2007, 190, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Ang II-stimulated superoxide production is mediated via phospholipase D in human vascular smooth muscle cells. Hypertens. 1999, 34, 976–982. [Google Scholar] [CrossRef]
- Széles, L.; Töröcsik, D.; Nagy, L. PPARgamma in immunity and inflammation: Cell types and diseases. Biochim. Biophys. Acta 2007, 1771, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A new nomenclature for int-1 and related genes: The Wnt gene family. Cell 1991, 64, 231. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Dev. Camb. Engl. 2004, 131, 1663–1677. [Google Scholar] [CrossRef]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, M.; Jho, E. Wnt/β-catenin signalling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.P.; Kühl, M. An updated overview on Wnt signaling pathways: A prelude for more. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; De Aguiar, R.B.; Naik, S.; Mani, S.; Ostadsharif, K.; Wencker, D.; Sotoudeh, M.; Malekzadeh, R.; Sherwin, R.S.; Mani, A. LRP6 enhances glucose metabolism by promoting TCF7L2-dependent insulin receptor expression and IGF receptor stabilization in humans. Cell Metab. 2013, 17, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, K.; Srivastava, R.; Dong, C.; Go, G.-W.; Li, N.; Iwakiri, Y.; Mani, A. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 2015, 29, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Shi, Y.; Tang, S.-J. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J. Neuroimmune Pharmacol. 2012, 7, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.C.; Shukla, D.K.; Stebbins, G.T.; Skias, D.D.; Jeffery, D.R.; Stefoski, D.; Katsamakis, G.; Feinstein, D.L. A pilot test of pioglitazone as an add-on in patients with relapsing remitting multiple sclerosis. J. Neuroimmunol. 2009, 211, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.K.; Kaiser, C.C.; Stebbins, G.T.; Feinstein, D.L. Effects of pioglitazone on diffusion tensor imaging indices in multiple sclerosis patients. Neurosci. Lett. 2010, 472, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Schmidt, M.; Giese, T.; Sastre, M.; Knolle, P.; Klockgether, T.; Heneka, M.T. Proinflammatory stimulation and pioglitazone treatment regulate peroxisome proliferator-activated receptor gamma levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J. Immunol. 1950 2005, 175, 4948–4955. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Beal, M.F.; Kiaei, M. Preferential PPAR-α activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Guillevin, R.; Vallée, J.-N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev. Neurosci. 2018, 29, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Interactions between PPAR Gamma and the Canonical Wnt/Beta-Catenin Pathway in Type 2 Diabetes and Colon Cancer. PPAR Res. 2017, 2017, 5879090. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Thermodynamics in cancers: Opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin. Transl. Med. 2017, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int. J. Mol. Sci. 2017, 18, 2537. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Schussler, O.; Claes, V.; Vallée, A. The Myofibroblast: TGFβ-1, A Conductor which Plays a Key Role in Fibrosis by Regulating the Balance between PPARγ and the Canonical WNT Pathway. Nucl. Recept. Res. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin. 2017, 49, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Reprogramming energetic metabolism in Alzheimer’s disease. Life Sci. 2018, 193, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J. Mol. Neurosci. 2017, 62, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. PPARγ agonists: Potential treatments for exudative age-related macular degeneration. Life Sci. 2017, 188, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Vallée, A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; D’Urso, M.C.; di Blasio, G.; Brignone, M.S.; De Simone, R.; Minghetti, L. Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain. Behav. Immun. 2016, 55, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Jansson, E.A.; Are, A.; Greicius, G.; Kuo, I.-C.; Kelly, D.; Arulampalam, V.; Pettersson, S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Drygiannakis, I.; Valatas, V.; Sfakianaki, O.; Bourikas, L.; Manousou, P.; Kambas, K.; Ritis, K.; Kolios, G.; Kouroumalis, E. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: Implication in intestinal fibrosis. J. Crohns Colitis 2013, 7, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Regulation of PPARgamma activity during adipogenesis. Int. J. Obes. 2005, 29 (Suppl. 1), S13–S16. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.-I.; Kulkarni, A.; Woeller, C.F.; Phipps, R.P.; Sime, P.J.; Hindman, H.B.; Huxlin, K.R. Inhibitory effects of PPARγ ligands on TGF-β1-induced corneal myofibroblast transformation. Am. J. Pathol. 2014, 184, 1429–1445. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mundra, V.; Mahato, R.I. Nanomedicines of Hedgehog inhibitor and PPAR-γ agonist for treating liver fibrosis. Pharm. Res. 2014, 31, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, S.H.; Lee, Y.J.; Kang, E.S.; Lee, B.-W.; Cha, B.S.; Kim, J.W.; Song, D.H.; Lee, H.C. Transcription factor Snail is a novel regulator of adipocyte differentiation via inhibiting the expression of peroxisome proliferator-activated receptor γ. Cell. Mol. Life Sci. 2013, 70, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, Z.; Li, F.; Lu, W.; Wang, J.; Guo, C. The improving effects on hepatic fibrosis of interferon-γ liposomes targeted to hepatic stellate cells. Nanotechnology 2012, 23, 265101. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, S.R. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J. Biol. Chem. 2004, 279, 45020–45027. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Niu, M.; Zhai, X.; Zhou, Q.; Zhou, Y. β-Catenin pathway is required for TGF-β1 inhibition of PPARγ expression in cultured hepatic stellate cells. Pharmacol. Res. 2012, 66, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Segel, M.J.; Izbicki, G.; Cohen, P.Y.; Or, R.; Christensen, T.G.; Wallach-Dayan, S.B.; Breuer, R. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1255–L1262. [Google Scholar] [CrossRef] [PubMed]
- Shim, C.Y.; Song, B.-W.; Cha, M.-J.; Hwang, K.-C.; Park, S.; Hong, G.-R.; Kang, S.-M.; Lee, J.E.; Ha, J.-W.; Chung, N. Combination of a peroxisome proliferator-activated receptor-gamma agonist and an angiotensin II receptor blocker attenuates myocardial fibrosis and dysfunction in type 2 diabetic rats. J. Diabetes Investig. 2014, 5, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-F.; Sun, Y.-Y.; Bao, J.; Chen, X.; Li, Y.-H.; Yang, Y.; Zhang, L.; Huang, C.; Wu, B.-M.; Meng, X.-M.; et al. Functional role of PPAR-γ on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci. Rep. 2017, 7, 12671. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Carson, D.A. Repression of beta-catenin signaling by PPAR gamma ligands. Eur. J. Pharmacol. 2010, 636, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Kouzmenko, A.P.; Kato, S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009, 5, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Elbrecht, A.; Chen, Y.; Cullinan, C.A.; Hayes, N.; Leibowitz, M.D.; Moller, D.E.; Berger, J. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem. Biophys. Res. Commun. 1996, 224, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed]
- Moldes, M.; Zuo, Y.; Morrison, R.F.; Silva, D.; Park, B.-H.; Liu, J.; Farmer, S.R. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem. J. 2003, 376, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Akinyeke, T.O.; Stewart, L.V. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARγ-independent mechanism. Cancer Biol. Ther. 2011, 11, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Eliasson, B.; Smith, U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: A link with osteogenesis. Diabetologia 2010, 53, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Rahman, N.; Kim, Y.-S. Wnt/β-catenin signaling plays a distinct role in methyl gallate-mediated inhibition of adipogenesis. Biochem. Biophys. Res. Commun. 2016, 479, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Kudo, H.; Wakabayashi, K.; Tanaka, T.; Nonaka, A.; Uchida, A.; Tsutsumi, S.; Sakakibara, I.; Naito, M.; Osborne, T.F.; et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.F.; Daviaud, D.; Pradère, J.P.; Grès, S.; Guigné, C.; Wabitsch, M.; Chun, J.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor gamma2. J. Biol. Chem. 2005, 280, 14656–14662. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.M.; McLennan, S.V.; Song, W.W.; Lo, L.W.-Y.; Bonner, J.G.; Williams, P.F.; Twigg, S.M. Connective tissue growth factor inhibits adipocyte differentiation. Am. J. Physiol. Cell Physiol. 2008, 295, C740–C751. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Nakashima, T.; Kawakami, A.; Miyashita, T.; Tanaka, F.; Ida, H.; Migita, K.; Origuchi, T.; Eguchi, K. Cytokines regulate fibroblast-like synovial cell differentiation to adipocyte-like cells. Rheumatology 2004, 43, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoé, K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J. Neuroimmunol. 2001, 116, 40–48. [Google Scholar] [CrossRef]
- Diab, A.; Deng, C.; Smith, J.D.; Hussain, R.Z.; Phanavanh, B.; Lovett-Racke, A.E.; Drew, P.D.; Racke, M.K. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Bright, J.J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002, 3, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G.; Shao, G.; Heyman, R.A. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimers: Intermolecular synergy requires only the PPARgamma hormone-dependent activation function. Mol. Cell. Biol. 1998, 18, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.; Kurokawa, R.; Nolte, R.T.; Wisely, G.B.; McInerney, E.M.; Rose, D.W.; Milburn, M.V.; Rosenfeld, M.G.; Glass, C.K. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature 1998, 395, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Jarjour, A.A.; Oumesmar, B.N.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Baron-Van Evercooren, A.; et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat. Neurosci. 2011, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Benedusi, V.; Martorana, F.; Brambilla, L.; Maggi, A.; Rossi, D. The peroxisome proliferator-activated receptor γ (PPARγ) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis. J. Biol. Chem. 2012, 287, 35899–35911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, P.D.; Storer, P.D.; Xu, J.; Chavis, J.A. Hormone regulation of microglial cell activation: Relevance to multiple sclerosis. Brain Res. Brain Res. Rev. 2005, 48, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Drew, P.D.; Xu, J.; Racke, M.K. PPAR-gamma: Therapeutic Potential for Multiple Sclerosis. PPAR Res. 2008, 2008, 627463. [Google Scholar] [CrossRef] [PubMed]
- Duvanel, C.B.; Honegger, P.; Pershadsingh, H.; Feinstein, D.; Matthieu, J.-M. Inhibition of glial cell proinflammatory activities by peroxisome proliferator-activated receptor gamma agonist confers partial protection during antimyelin oligodendrocyte glycoprotein demyelination in vitro. J. Neurosci. Res. 2003, 71, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martínez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, I.; Singh, A.K. IL-4-induced peroxisome proliferator-activated receptor gamma activation inhibits NF-kappaB trans activation in central nervous system (CNS) glial cells and protects oligodendrocyte progenitors under neuroinflammatory disease conditions: Implication for CNS-demyelinating diseases. J. Immunol. 2006, 176, 4385–4398. [Google Scholar] [PubMed]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Xin, T.; Hunter, R.L.; Bing, G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J. Neuroinflamm. 2008, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Novak, N.; Beyer, M.; et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Unoda, K.; Doi, Y.; Nakajima, H.; Yamane, K.; Hosokawa, T.; Ishida, S.; Kimura, F.; Hanafusa, T. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 256, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Storer, P.D.; Xu, J.; Chavis, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. J. Neuroimmunol. 2005, 161, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists suppress the production of IL-12 family cytokines by activated glia. J. Immunol. 2007, 178, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Leung, E.; Phipps, R.P. Human B lymphocytes and B lymphomas express PPAR-gamma and are killed by PPAR-gamma agonists. Clin. Immunol. 2002, 103, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Stanislaus, R.; Gilg, A.G.; Barbosa, E.; Singh, I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J. Neurosci. Res. 2004, 77, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, S.; Merkler, D.; Schmitz, M.; Klöppner, S.; Schedensack, M.; Jeserich, G.; Althaus, H.H.; Brück, W. Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J. Neurosci. 2008, 28, 13609–13614. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Zehntner, S.P.; Kuhlmann, T.; Ludwin, S.K.; Owens, T.; Kennedy, T.E.; Bedell, B.J.; Antel, J.P. Statin therapy inhibits remyelination in the central nervous system. Am. J. Pathol. 2009, 174, 1880–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancy, S.P.J.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.M.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Kishioka, S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr. Opin. Pharmacol. 2012, 12, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.; Quaranta, V. Cadherin-bound beta-catenin feeds into the Wnt pathway upon adherens junctions dissociation: Evidence for an intersection between beta-catenin pools. PLoS ONE 2009, 4, e4580. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Padden, M.; Leech, S.; Craig, B.; Kirk, J.; Brankin, B.; McQuaid, S. Differences in expression of junctional adhesion molecule-A and beta-catenin in multiple sclerosis brain tissue: Increasing evidence for the role of tight junction pathology. Acta Neuropathol. 2007, 113, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Soundara Rajan, T.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. Moringin activates Wnt canonical pathway by inhibiting GSK3β in a mouse model of experimental autoimmune encephalomyelitis. Drug Des. Dev. Ther. 2016, 10, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; De Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Montaut, S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. An overview on neuroprotective effects of isothiocyanates for the treatment of neurodegenerative diseases. Fitoterapia 2015, 106, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N. Warburg effect hypothesis in autism Spectrum disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Lee, R.D.; Kang, S.-K.; Han, S.Y.; Park, K.L.; Yang, K.H.; Song, Y.S.; Park, H.J.; Lee, Y.M.; Yun, Y.P.; et al. Neuronal differentiation of embryonic midbrain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp. Cell Res. 2004, 297, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lan, F.; Yang, W.; Yang, Y.; Han, L.; Zhang, A.; Liu, J.; Zeng, H.; Jiang, T.; Pu, P.; et al. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010, 1366, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Goldbeter, A. Patterns of spatiotemporal organization in an allosteric enzyme model. Proc. Natl. Acad. Sci. USA 1973, 70, 3255–3259. [Google Scholar] [CrossRef] [PubMed]
- Cambron, M.; D’Haeseleer, M.; Laureys, G.; Clinckers, R.; Debruyne, J.; De Keyser, J. White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J. Cereb. Blood Flow Metab. 2012, 32, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Hübers, A.; Wahl, M.; Ziemann, U. Combined (1)H and (31)P spectroscopy provides new insights into the pathobiochemistry of brain damage in multiple sclerosis. NMR Biomed. 2011, 24, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, M.E.; Bø, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.G.; De Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 2009, 219, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Gebregiworgis, T.; Nielsen, H.H.; Massilamany, C.; Gangaplara, A.; Reddy, J.; Illes, Z.; Powers, R. A Urinary Metabolic Signature for Multiple Sclerosis and Neuromyelitis Optica. J. Proteome Res. 2016, 15, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Molenaar, R.J.; van der Pol, S.M.A.; van der Valk, P.; van Noorden, C.J.F.; de Vries, H.E.; van Horssen, J. Differential expression of glucose-metabolizing enzymes in multiple sclerosis lesions. Acta Neuropathol. Commun. 2015, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics profile of CD4(+) T cells in relapsing remitting multiple sclerosis subjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- De Riccardis, L.; Ferramosca, A.; Danieli, A.; Trianni, G.; Zara, V.; De Robertis, F.; Maffia, M. Metabolic response to glatiramer acetate therapy in multiple sclerosis patients. BBA Clin. 2016, 6, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, A.L.; Barks, S.K.; Duka, T.; Grossman, L.I.; Hof, P.R.; Sherwood, C.C. Aerobic glycolysis in the primate brain: Reconsidering the implications for growth and maintenance. Brain Struct. Funct. 2014, 219, 1149–1167. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, E.R.; Pasquini, J.M.; Soto, E.F. Oxidative damage to proteins and lipids of CNS myelin produced by in vitro generated reactive oxygen species. J. Neurosci. Res. 1995, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.A.; Hermfisse, U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. FASEB J. 1997, 11, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, P.; Fernandez, E.; Almeida, A.; Bolaños, J.P. Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. 2012, 19, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.-A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012, 106, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, L.K.; Araújo, W.L.; Strokina, A.A.; Fernie, A.R.; Bettendorff, L.; Bunik, V.I. Consequences of the α-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 5895–5906. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, C.; Van Hecke, P.; Vandenberghe, R.; Van Oostende, S.; Dupont, P.; Demaerel, P.; Bormans, G.; Carton, H. Positron emission tomography, magnetic resonance imaging and proton NMR spectroscopy of white matter in multiple sclerosis. Mult. Scler. J. 1997, 3, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Simone, I.L.; Tortorella, C.; Federico, F.; Liguori, M.; Lucivero, V.; Giannini, P.; Carrara, D.; Bellacosa, A.; Livrea, P. Axonal damage in multiple sclerosis plaques: A combined magnetic resonance imaging and 1H-magnetic resonance spectroscopy study. J. Neurol. Sci. 2001, 182, 143–150. [Google Scholar] [CrossRef]
- Amorini, A.M.; Nociti, V.; Petzold, A.; Gasperini, C.; Quartuccio, E.; Lazzarino, G.; Di Pietro, V.; Belli, A.; Signoretti, S.; Vagnozzi, R.; et al. Serum lactate as a novel potential biomarker in multiple sclerosis. Biochim. Biophys. Acta 2014, 1842, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Nijland, P.G.; Balk, L.J.; Amorini, A.M.; Lazzarino, G.; Wattjes, M.P.; Gasperini, C.; van der Valk, P.; Tavazzi, B.; Lazzarino, G.; et al. Visual pathway neurodegeneration winged by mitochondrial dysfunction. Ann. Clin. Transl. Neurol. 2015, 2, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Schocke, M.F.H.; Berger, T.; Felber, S.R.; Wolf, C.; Deisenhammer, F.; Kremser, C.; Seppi, K.; Aichner, F.T. Serial contrast-enhanced magnetic resonance imaging and spectroscopic imaging of acute multiple sclerosis lesions under high-dose methylprednisolone therapy. NeuroImage 2003, 20, 1253–1263. [Google Scholar] [CrossRef]
- Lutz, N.W.; Viola, A.; Malikova, I.; Confort-Gouny, S.; Audoin, B.; Ranjeva, J.-P.; Pelletier, J.; Cozzone, P.J. Inflammatory multiple-sclerosis plaques generate characteristic metabolic profiles in cerebrospinal fluid. PLoS ONE 2007, 2, e595. [Google Scholar] [CrossRef] [PubMed]
- Regenold, W.T.; Phatak, P.; Makley, M.J.; Stone, R.D.; Kling, M.A. Cerebrospinal fluid evidence of increased extra-mitochondrial glucose metabolism implicates mitochondrial dysfunction in multiple sclerosis disease progression. J. Neurol. Sci. 2008, 275, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Simone, I.L.; Federico, F.; Trojano, M.; Tortorella, C.; Liguori, M.; Giannini, P.; Picciola, E.; Natile, G.; Livrea, P. High resolution proton MR spectroscopy of cerebrospinal fluid in MS patients. Comparison with biochemical changes in demyelinating plaques. J. Neurol. Sci. 1996, 144, 182–190. [Google Scholar] [CrossRef]
- Hogenesch, J.B.; Gu, Y.Z.; Jain, S.; Bradfield, C.A. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc. Natl. Acad. Sci. USA 1998, 95, 5474–5479. [Google Scholar] [CrossRef] [PubMed]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Schibler, U.; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar] [CrossRef]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- De Somma, E.; Jain, R.W.; Poon, K.W.C.; Tresidder, K.A.; Segal, J.P.; Ghasemlou, N. Chronobiological regulation of psychosocial and physiological outcomes in multiple sclerosis. Neurosci. Biobehav. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Åkerstedt, T.; Hillert, J.; Olsson, T.; Alfredsson, L. Shift work at young age is associated with increased risk for multiple sclerosis. Ann. Neurol. 2011, 70, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Åkerstedt, T.; Olsson, T.; Alfredsson, L. Shift work influences multiple sclerosis risk. Mult. Scler. J. 2015, 21, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Haspel, J.A.; Chettimada, S.; Shaik, R.S.; Chu, J.-H.; Raby, B.A.; Cernadas, M.; Carey, V.; Process, V.; Hunninghake, G.M.; Ifedigbo, E.; et al. Circadian rhythm reprogramming during lung inflammation. Nat. Commun. 2014, 5, 4753. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hsuchou, H.; He, Y.; Kastin, A.J.; Wang, Y.; Pan, W. Sleep restriction impairs blood-brain barrier function. J. Neurosci. 2014, 34, 14697–14706. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, Z.; Tokgöz, S.; Gökbel, H.; Okudan, N.; Uğuz, F.; Yilmaz, G. The association of nocturnal serum melatonin levels with major depression in patients with acute multiple sclerosis. Psychiatry Res. 2008, 161, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Marck, C.H.; Neate, S.L.; Taylor, K.L.; Weiland, T.J.; Jelinek, G.A. Prevalence of Comorbidities, Overweight and Obesity in an International Sample of People with Multiple Sclerosis and Associations with Modifiable Lifestyle Factors. PLoS ONE 2016, 11, e0148573. [Google Scholar] [CrossRef] [PubMed]
- Lunde, H.M.B.; Bjorvatn, B.; Myhr, K.-M.; Bø, L. Clinical assessment and management of sleep disorders in multiple sclerosis: A literature review. Acta Neurol. Scand. 2013, 127, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.W.; Dugovic, C.; Zee, P.C. Current understanding of the circadian clock and the clinical implications for neurological disorders. Arch. Neurol. 2001, 58, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Attarian, H.P.; Brown, K.M.; Duntley, S.P.; Carter, J.D.; Cross, A.H. The relationship of sleep disturbances and fatigue in multiple sclerosis. Arch. Neurol. 2004, 61, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.R.; Toghianifar, N.; Etemadifar, M.; Haghighi, S.; Maghzi, A.H.; Akbari, M. Circadian rhythm sleep disorders in patients with multiple sclerosis and its association with fatigue: A case-control study. J. Res. Med. Sci. 2013, 18, S71–S73. [Google Scholar] [PubMed]
- Ayache, S.S.; Chalah, M.A. Fatigue in multiple sclerosis—Insights into evaluation and management. Neurophysiol. Clin. Clin. Neurophysiol. 2017, 47, 139–171. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, N.; Sakurai, T. Role of orexin in modulating arousal, feeding, and motivation. Front. Behav. Neurosci. 2013, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Kiyashchenko, L.I.; Mileykovskiy, B.Y.; Maidment, N.; Lam, H.A.; Wu, M.-F.; John, J.; Peever, J.; Siegel, J.M. Release of hypocretin (orexin) during waking and sleep states. J. Neurosci. 2002, 22, 5282–5286. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Kanbayashi, T.; Mezaki, T.; Iseki, K.; Matsubayashi, J.; Murakami, G.; Matsui, M.; Shimizu, T.; Shibasaki, H. Low CSF hypocretin-1/orexin-A associated with hypersomnia secondary to hypothalamic lesion in a case of multiple sclerosis. J. Neurol. 2004, 251, 885–886. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Zhu, X.; Leinninger, G.M.; Levasseur, P.R.; Braun, T.P.; Myers, M.G.; Marks, D.L. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J. Neurosci. 2011, 31, 11376–11386. [Google Scholar] [CrossRef] [PubMed]
- Pokryszko-Dragan, A.; Frydecka, I.; Kosmaczewska, A.; Ciszak, L.; Bilińska, M.; Gruszka, E.; Podemski, R.; Frydecka, D. Stimulated peripheral production of interferon-gamma is related to fatigue and depression in multiple sclerosis. Clin. Neurol. Neurosurg. 2012, 114, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Boddum, K.; Hansen, M.H.; Jennum, P.J.; Kornum, B.R. Cerebrospinal Fluid Hypocretin-1 (Orexin-A) Level Fluctuates with Season and Correlates with Day Length. PLoS ONE 2016, 11, e0151288. [Google Scholar] [CrossRef] [PubMed]
- Taphoorn, M.J.; van Someren, E.; Snoek, F.J.; Strijers, R.L.; Swaab, D.F.; Visscher, F.; de Waal, L.P.; Polman, C.H. Fatigue, sleep disturbances and circadian rhythm in multiple sclerosis. J. Neurol. 1993, 240, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Farez, M.F.; Mascanfroni, I.D.; Méndez-Huergo, S.P.; Yeste, A.; Murugaiyan, G.; Garo, L.P.; Balbuena Aguirre, M.E.; Patel, B.; Ysrraelit, M.C.; Zhu, C.; et al. Melatonin Contributes to the Seasonality of Multiple Sclerosis Relapses. Cell 2015, 162, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Torkildsen, O.; Aarseth, J.; Benjaminsen, E.; Celius, E.; Holmøy, T.; Kampman, M.T.; Løken-Amsrud, K.; Midgard, R.; Myhr, K.-M.; Riise, T.; et al. Month of birth and risk of multiple sclerosis: Confounding and adjustments. Ann. Clin. Transl. Neurol. 2014, 1, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, J.D.; Case, L.K.; Krementsov, D.N.; Raza, A.; Bartiss, R.; Teuscher, C. Modeling month-season of birth as a risk factor in mouse models of chronic disease: From multiple sclerosis to autoimmune encephalomyelitis. FASEB J. 2017, 31, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.K.; Tesfagiorgis, Y.; Jain, R.W.; Craig, H.C.; Kerfoot, S.M. Meningeal Infiltration of the Spinal Cord by Non-Classically Activated B Cells is Associated with Chronic Disease Course in a Spontaneous B Cell-Dependent Model of CNS Autoimmune Disease. Front. Immunol. 2015, 6, 470. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Rollins, D.; Ruhn, K.A.; Stubblefield, J.J.; Green, C.B.; Kashiwada, M.; Rothman, P.B.; Takahashi, J.S.; Hooper, L.V. TH17 cell differentiation is regulated by the circadian clock. Science 2013, 342, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.L. Inhibition of growth and differentiation of osteoprogenitors in mouse bone marrow stromal cell cultures by increased donor age and glucocorticoid treatment. Bone 2004, 35, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Soták, M.; Sumová, A.; Pácha, J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann. Med. 2014, 46, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; He, D.; Zhang, W.; Walton, R.G. Trends in prevalence, awareness, management, and control of hypertension among United States adults, 1999 to 2010. J. Am. Coll. Cardiol. 2012, 60, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Yasuniwa, Y.; Izumi, H.; Wang, K.-Y.; Shimajiri, S.; Sasaguri, Y.; Kawai, K.; Kasai, H.; Shimada, T.; Miyake, K.; Kashiwagi, E.; et al. Circadian disruption accelerates tumor growth and angio/stromagenesis through a Wnt signaling pathway. PLoS ONE 2010, 5, e15330. [Google Scholar] [CrossRef] [PubMed]
- Janich, P.; Pascual, G.; Merlos-Suárez, A.; Batlle, E.; Ripperger, J.; Albrecht, U.; Cheng, H.-Y.M.; Obrietan, K.; Di Croce, L.; Benitah, S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 2011, 480, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Lavtar, P.; Rudolf, G.; Maver, A.; Hodžić, A.; Starčević Čizmarević, N.; Živković, M.; Šega Jazbec, S.; Klemenc Ketiš, Z.; Kapović, M.; Dinčić, E.; et al. Association of circadian rhythm genes ARNTL/BMAL1 and CLOCK with multiple sclerosis. PLoS ONE 2018, 13, e0190601. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Chen, Y.; Li, X.; Zhao, Q.; Tan, Z. Over-expression of circadian clock gene Bmal1 affects proliferation and the canonical Wnt pathway in NIH-3T3 cells. Cell Biochem. Funct. 2013, 31, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wood, P.A.; Ansell, C.M.; Ohmori, M.; Oh, E.-Y.; Xiong, Y.; Berger, F.G.; Peña, M.M.O.; Hrushesky, W.J.M. Beta-catenin induces beta-TrCP-mediated PER2 degradation altering circadian clock gene expression in intestinal mucosa of ApcMin/+ mice. J. Biochem. 2009, 145, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Duffield, G.E.; Best, J.D.; Meurers, B.H.; Bittner, A.; Loros, J.J.; Dunlap, J.C. Circadian programs of transcriptional activation, signaling, and protein turnover revealed by microarray analysis of mammalian cells. Curr. Biol. 2002, 12, 551–557. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Downes, M.; Yu, R.T.; Bookout, A.L.; He, W.; Straume, M.; Mangelsdorf, D.J.; Evans, R.M. Nuclear receptor expression links the circadian clock to metabolism. Cell 2006, 126, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Jia, Z.; Aoyagi, T.; McClain, D.; Mortensen, R.M.; Yang, T. Systemic PPARγ deletion impairs circadian rhythms of behavior and metabolism. PLoS ONE 2012, 7, e38117. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-M.; Zhao, Y.-X.; Zhang, S.; Liu, G.-D.; Kang, W.-Y.; Tang, H.-D.; Ding, J.-Q.; Chen, S.-D. PPARgamma agonist curcumin reduces the amyloid-beta-stimulated inflammatory responses in primary astrocytes. J. Alzheimers Dis. 2010, 20, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, C.; Dubois, G.; Duguay, Y.; Helledie, T.; Vu-Dac, N.; Gervois, P.; Soncin, F.; Mandrup, S.; Fruchart, J.-C.; Fruchart-Najib, J.; et al. The orphan nuclear receptor Rev-Erbalpha is a peroxisome proliferator-activated receptor (PPAR) gamma target gene and promotes PPARgamma-induced adipocyte differentiation. J. Biol. Chem. 2003, 278, 37672–37680. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Douris, N.; Kojima, S.; Strayer, C.A.; Fogerty, J.; Lourim, D.; Keller, S.R.; Besharse, J.C. Loss of Nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 9888–9893. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, G. PPARs Integrate the Mammalian Clock and Energy Metabolism. PPAR Res. 2014, 2014, 653017. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B.; Bishop-Bailey, D.; Estrada-Hernandez, T.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Z.; Zhang, K.; Xue, Z.; Li, Y.; Zhang, Z.; Zhang, L.; Gu, C.; Zhang, Q.; Hao, J.; et al. Arctigenin Suppress Th17 Cells and Ameliorates Experimental Autoimmune Encephalomyelitis Through AMPK and PPAR-γ/ROR-γt Signaling. Mol. Neurobiol. 2016, 53, 5356–5366. [Google Scholar] [CrossRef] [PubMed]
- Lochner, M.; Peduto, L.; Cherrier, M.; Sawa, S.; Langa, F.; Varona, R.; Riethmacher, D.; Si-Tahar, M.; Di Santo, J.P.; Eberl, G. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J. Exp. Med. 2008, 205, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Cermakian, N.; Lange, T.; Golombek, D.; Sarkar, D.; Nakao, A.; Shibata, S.; Mazzoccoli, G. Crosstalk between the circadian clock circuitry and the immune system. Chronobiol. Int. 2013, 30, 870–888. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Sulli, A.; Pincus, T. Circadian use of glucocorticoids in rheumatoid arthritis. Neuroimmunomodulation 2015, 22, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Schultheiss, T.; Schneider, H.; Schrempf, W.; Reichmann, H.; Ziemssen, T. Circadian cortisol, depressive symptoms and neurological impairment in early multiple sclerosis. Psychoneuroendocrinology 2011, 36, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Wipfler, P.; Heikkinen, A.; Harrer, A.; Pilz, G.; Kunz, A.; Golaszewski, S.M.; Reuss, R.; Oschmann, P.; Kraus, J. Circadian rhythmicity of inflammatory serum parameters: A neglected issue in the search of biomarkers in multiple sclerosis. J. Neurol. 2013, 260, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.H.; Moss-Morris, R.; Liossi, C.; Schlotz, W. Circadian cortisol and fatigue severity in relapsing-remitting multiple sclerosis. Psychoneuroendocrinology 2015, 56, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Melief, J.; de Wit, S.J.; van Eden, C.G.; Teunissen, C.; Hamann, J.; Uitdehaag, B.M.; Swaab, D.; Huitinga, I. HPA axis activity in multiple sclerosis correlates with disease severity, lesion type and gene expression in normal-appearing white matter. Acta Neuropathol. 2013, 126, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.; MacPhee, I.; Antoni, F. The role of the neuroendocrine system in determining genetic susceptibility to experimental allergic encephalomyelitis in the rat. Immunology 1990, 70, 1–5. [Google Scholar] [PubMed]
- Gold, S.M.; Raji, A.; Huitinga, I.; Wiedemann, K.; Schulz, K.-H.; Heesen, C. Hypothalamo-pituitary-adrenal axis activity predicts disease progression in multiple sclerosis. J. Neuroimmunol. 2005, 165, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Heidbrink, C.; Häusler, S.F.M.; Buttmann, M.; Ossadnik, M.; Strik, H.M.; Keller, A.; Buck, D.; Verbraak, E.; van Meurs, M.; Krockenberger, M.; et al. Reduced cortisol levels in cerebrospinal fluid and differential distribution of 11beta-hydroxysteroid dehydrogenases in multiple sclerosis: Implications for lesion pathogenesis. Brain. Behav. Immun. 2010, 24, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Van den Brandt, J.; Lühder, F.; McPherson, K.G.; de Graaf, K.L.; Tischner, D.; Wiehr, S.; Herrmann, T.; Weissert, R.; Gold, R.; Reichardt, H.M. Enhanced glucocorticoid receptor signaling in T cells impacts thymocyte apoptosis and adaptive immune responses. Am. J. Pathol. 2007, 170, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.-A.; Rivest, S. The HPA—Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front. Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Van Winsen, L.M.L.; Muris, D.F.R.; Polman, C.H.; Dijkstra, C.D.; van den Berg, T.K.; Uitdehaag, B.M.J. Sensitivity to glucocorticoids is decreased in relapsing remitting multiple sclerosis. J. Clin. Endocrinol. Metab. 2005, 90, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.; Park, N.; Cho, S.; Kim, K. Glucocorticoid-mediated Period2 induction delays the phase of circadian rhythm. Nucleic Acids Res. 2013, 41, 6161–6174. [Google Scholar] [CrossRef] [PubMed]
- So, A.Y.-L.; Bernal, T.U.; Pillsbury, M.L.; Yamamoto, K.R.; Feldman, B.J. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 17582–17587. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Xie, D.; Choudhary, V.; Seremwe, M.; Tsai, Y.-Y.; Olala, L.; Chen, X.; Bollag, W.B. The effect of pioglitazone on aldosterone and cortisol production in HAC15 human adrenocortical carcinoma cells. Mol. Cell. Endocrinol. 2014, 394, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Csernus, V.; Mess, B. Biorhythms and pineal gland. Neuro Endocrinol. Lett. 2003, 24, 404–411. [Google Scholar] [PubMed]
- Mauriz, J.L.; Collado, P.S.; Veneroso, C.; Reiter, R.J.; González-Gallego, J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: Recent insights and new perspectives. J. Pineal Res. 2013, 54, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.J.; Eastman, C.I. Melatonin in the afternoons of a gradually advancing sleep schedule enhances the circadian rhythm phase advance. Psychopharmacology 2013, 225, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Farez, M.F.; Calandri, I.L.; Correale, J.; Quintana, F.J. Anti-inflammatory effects of melatonin in multiple sclerosis. BioEssays 2016, 38, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Sandyk, R.; Awerbuch, G.I. Nocturnal plasma melatonin and alpha-melanocyte stimulating hormone levels during exacerbation of multiple sclerosis. Int. J. Neurosci. 1992, 67, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.-X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.R.; González-Yanes, C.; Maldonado, M.D. The role of melatonin in the cells of the innate immunity: A review. J. Pineal Res. 2013, 55, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-J.; Huang, S.-H.; Chen, J.-W.; Wang, K.-C.; Yang, Y.-R.; Liu, P.-F.; Lin, G.-J.; Sytwu, H.-K. Melatonin enhances interleukin-10 expression and suppresses chemotaxis to inhibit inflammation in situ and reduce the severity of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2016, 31, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Markowska, M.; Bialecka, B.; Ciechanowska, M.; Koter, Z.; Laskowska, H.; Karkucinska-Wieckowska, A.; Skwarlo-Sonta, K. Effect of immunization on nocturnal NAT activity in chicken pineal gland. Neuro Endocrinol. Lett. 2000, 21, 367–373. [Google Scholar] [PubMed]
- Fernandes, P.A.C.M.; Cecon, E.; Markus, R.P.; Ferreira, Z.S. Effect of TNF-alpha on the melatonin synthetic pathway in the rat pineal gland: Basis for a “feedback” of the immune response on circadian timing. J. Pineal Res. 2006, 41, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.P.; Bochenek, J.; Król, K.; Krawczyńska, A.; Antushevich, H.; Pawlina, B.; Herman, A.; Romanowicz, K.; Tomaszewska-Zaremba, D. Central Interleukin-1β Suppresses the Nocturnal Secretion of Melatonin. Mediators Inflamm. 2016, 2016, 2589483. [Google Scholar] [CrossRef] [PubMed]
- Pontes, G.N.; Cardoso, E.C.; Carneiro-Sampaio, M.M.S.; Markus, R.P. Pineal melatonin and the innate immune response: The TNF-alpha increase after cesarean section suppresses nocturnal melatonin production. J. Pineal Res. 2007, 43, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Kallaur, A.P.; Oliveira, S.R.; Simão, A.N.C.; Alfieri, D.F.; Flauzino, T.; Lopes, J.; de Carvalho Jennings Pereira, W.L.; de Meleck Proença, C.; Borelli, S.D.; Kaimen-Maciel, D.R.; et al. Cytokine Profile in Patients with Progressive Multiple Sclerosis and Its Association with Disease Progression and Disability. Mol. Neurobiol. 2017, 54, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Sánchez, N.; Cruz-Chamorro, I.; López-González, A.; Utrilla, J.C.; Fernández-Santos, J.M.; Martínez-López, A.; Lardone, P.J.; Guerrero, J.M.; Carrillo-Vico, A. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain. Behav. Immun. 2015, 50, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Hilliard, B.; Ventura, E.; Rostami, A. Luzindole, a melatonin receptor antagonist, suppresses experimental autoimmune encephalomyelitis. Pathobiology 1997, 65, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.C.; Ahn, M.; Kim, Y.S.; Moon, C.; Lee, Y.; Wie, M.B.; Lee, Y.J.; Shin, T. Melatonin ameliorates autoimmune encephalomyelitis through suppression of intercellular adhesion molecule-1. J. Vet. Sci. 2001, 2, 85–89. [Google Scholar] [PubMed]
- Giese, K.P. GSK-3: A key player in neurodegeneration and memory. IUBMB Life 2009, 61, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.B.; Frozza, R.L.; Horn, A.P.; Comiran, R.A.; Bernardi, A.; Campos, M.M.; Battastini, A.M.O.; Salbego, C. Amyloid-beta neurotoxicity in organotypic culture is attenuated by melatonin: Involvement of GSK-3beta, tau and neuroinflammation. J. Pineal Res. 2010, 48, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Guven, C.; Taskin, E.; Akcakaya, H. Melatonin Prevents Mitochondrial Damage Induced by Doxorubicin in Mouse Fibroblasts Through Ampk-Ppar Gamma-Dependent Mechanisms. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 438–446. [Google Scholar] [CrossRef]
- Kato, H.; Tanaka, G.; Masuda, S.; Ogasawara, J.; Sakurai, T.; Kizaki, T.; Ohno, H.; Izawa, T. Melatonin promotes adipogenesis and mitochondrial biogenesis in 3T3-L1 preadipocytes. J. Pineal Res. 2015, 59, 267–275. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Expression | Actions | Model | References |
|---|---|---|---|---|
| PPARγ | Agonists | Inhibition of NF-κB | EAE models | [114,115,116] |
| Decrease inflammation, permits remyelination | OLs models | [118,119] | ||
| Neuroprotection | EAE models | [121,122,123,124,125,126,127,128] | ||
| Th17 differentiation | Murine CD4+ T cells | [129] | ||
| Decrease IL-17 expression | EAE models | [130] | ||
| Decrease IL-1, IL-6 and COX2 | EAE models | [142] | ||
| Decrease β-catenin | EAE models | [142] | ||
| WNT | Overexpression | Chronic pain | EAE models | [68] |
| Impairs OPC differentiation | EAE models | [137] | ||
| Alteration of endothelial adherens | EAE models | [139,140] | ||
| Alteration of endothelial adherens | MS brain tissue | [141] | ||
| Aerobic Glycolysis | Activation | Neuronal cell death and astrocytic inflammation | EAE models | [166,167] |
| MS progression | Human models | [172,173] | ||
| Increased lactate production | Human models | [31,159] | ||
| Mitochondrial dysregulation | Human models | [175,176,177] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches. Int. J. Mol. Sci. 2018, 19, 1212. https://doi.org/10.3390/ijms19041212
Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches. International Journal of Molecular Sciences. 2018; 19(4):1212. https://doi.org/10.3390/ijms19041212
Chicago/Turabian StyleVallée, Alexandre, Yves Lecarpentier, Rémy Guillevin, and Jean-Noël Vallée. 2018. "Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches" International Journal of Molecular Sciences 19, no. 4: 1212. https://doi.org/10.3390/ijms19041212