The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
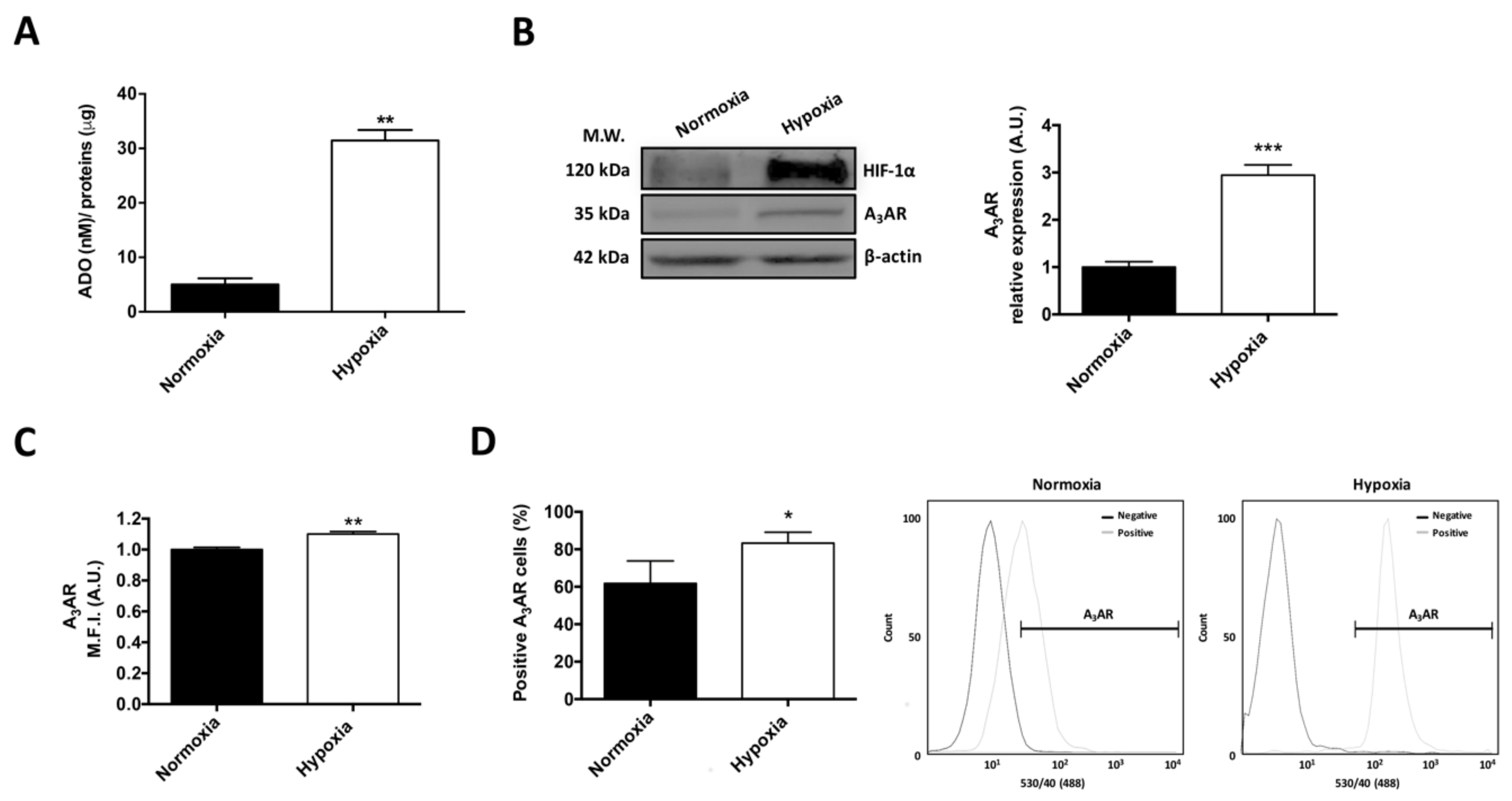
2.1. Extracellular Adenosine Concentration and A3 Adenosine Receptor Expression Increase under Hypoxia
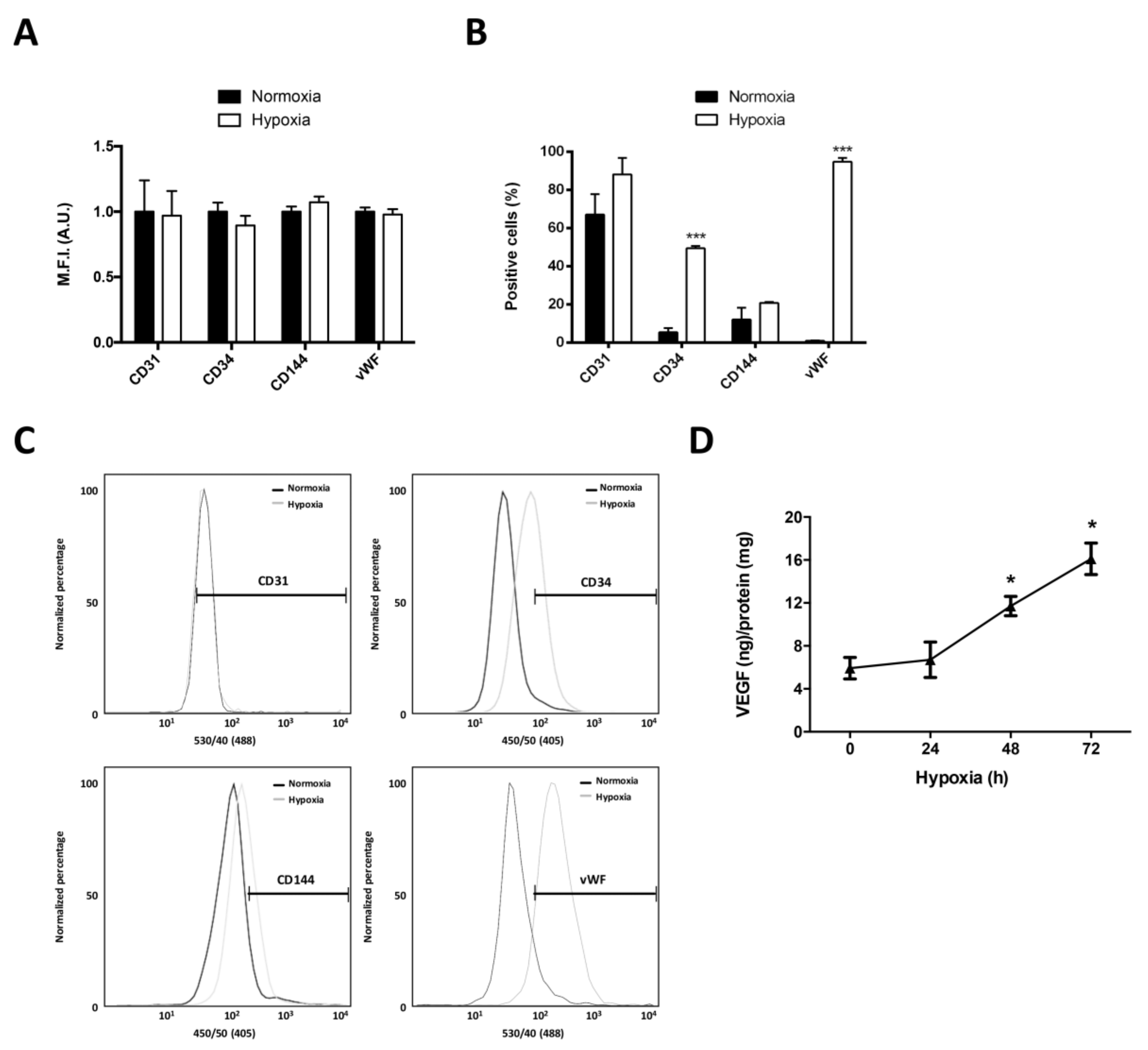
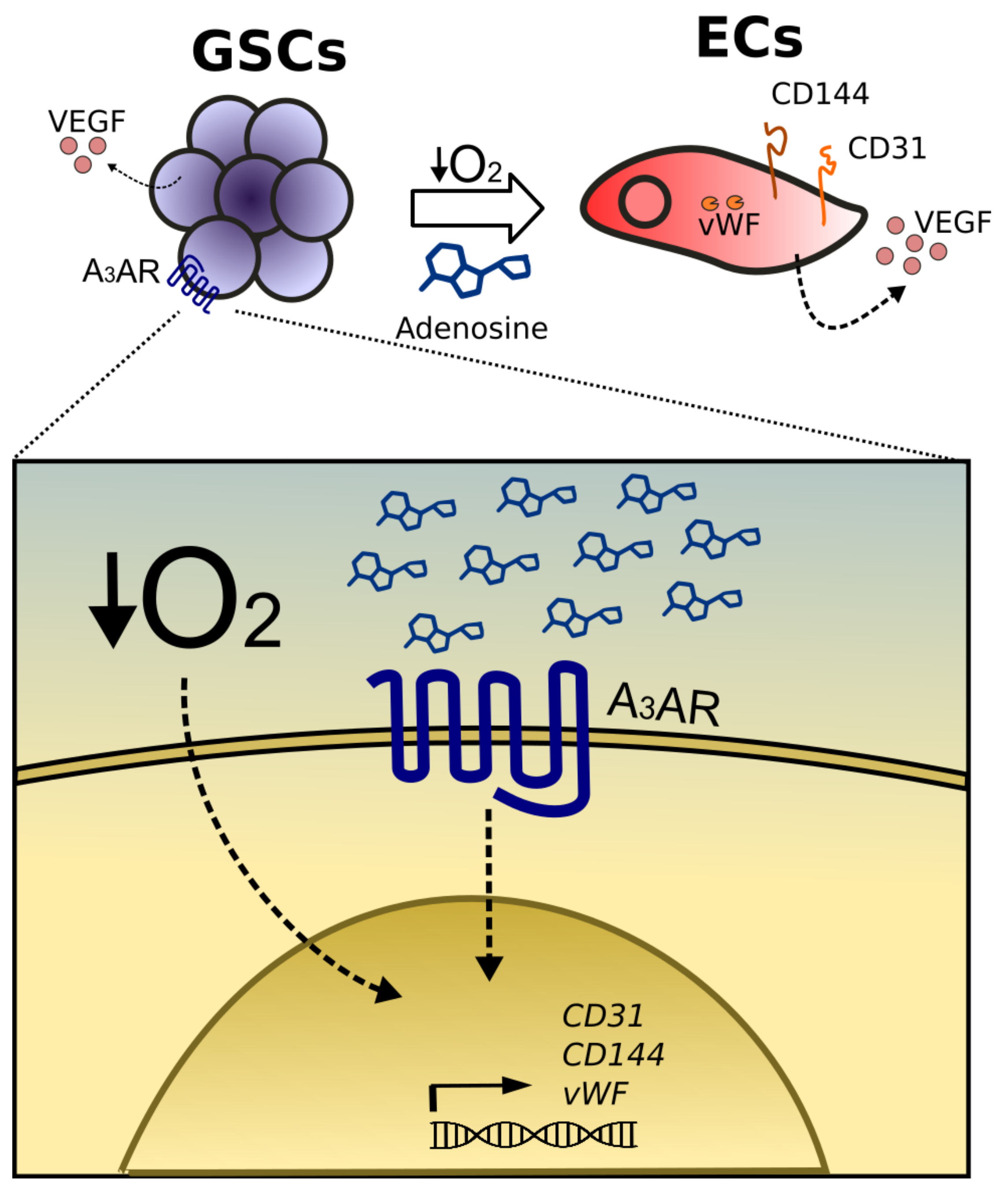
2.2. Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells Increases under Hypoxia
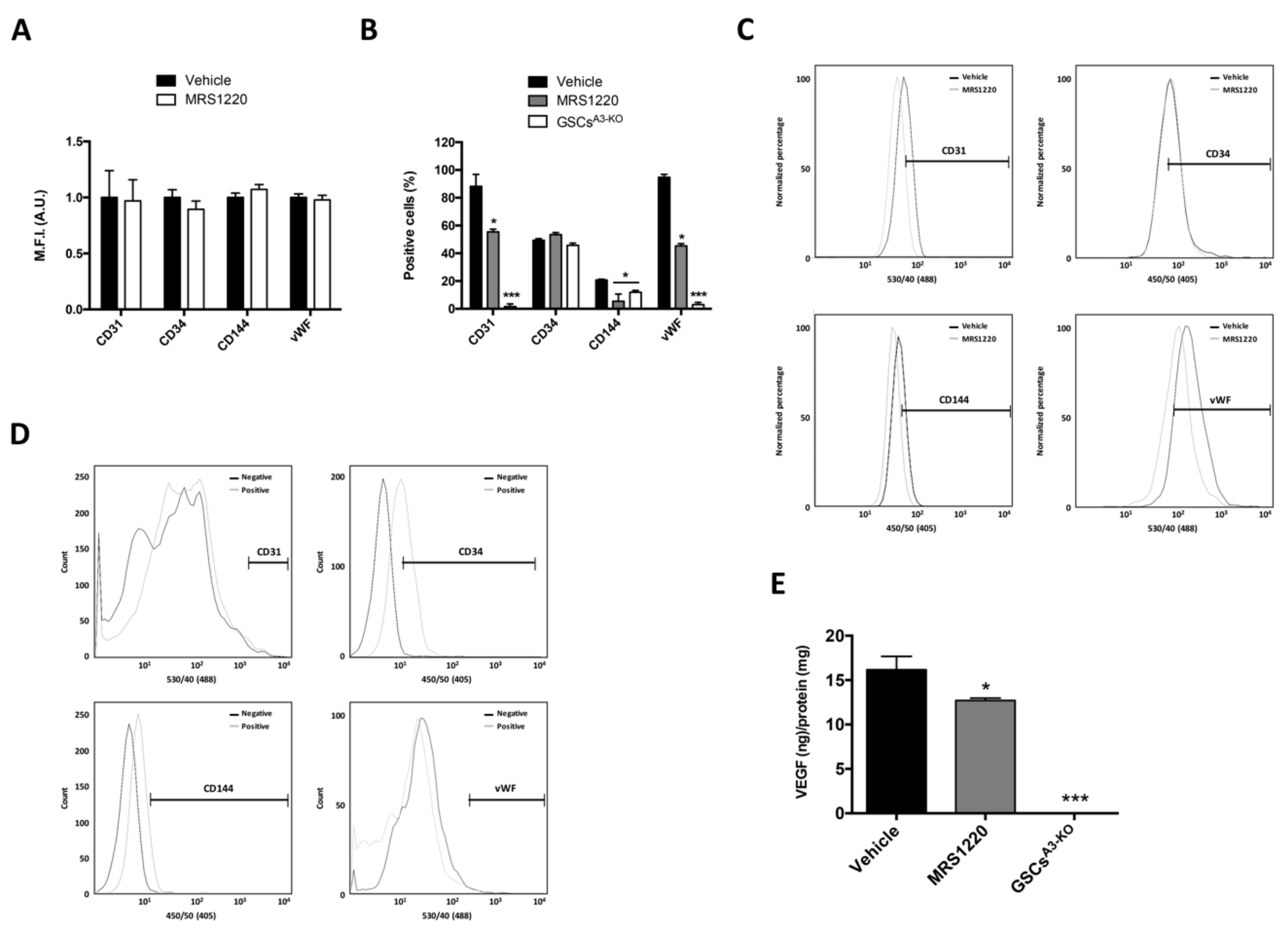
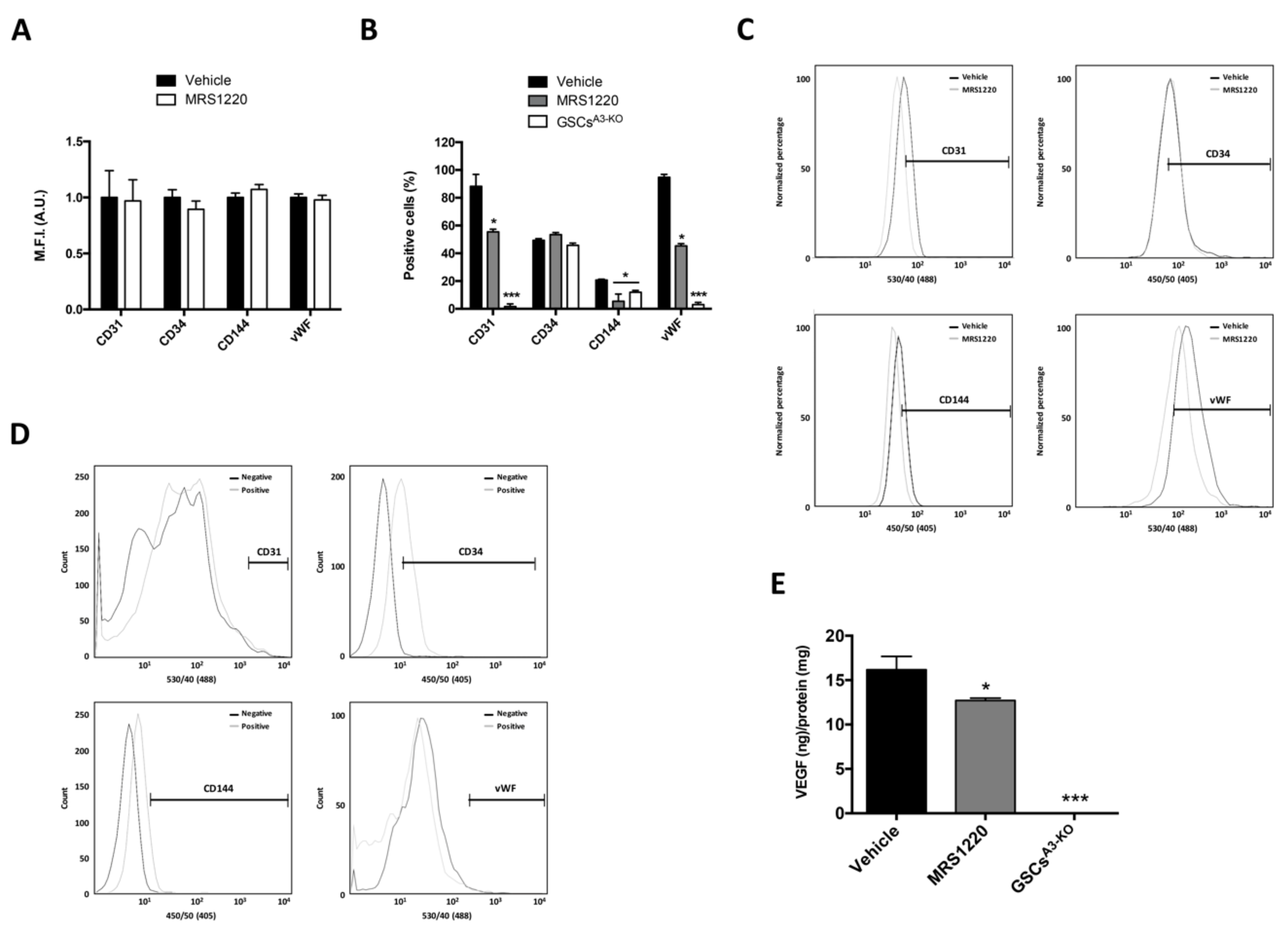
2.3. A3AR Blockade Decreases Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia
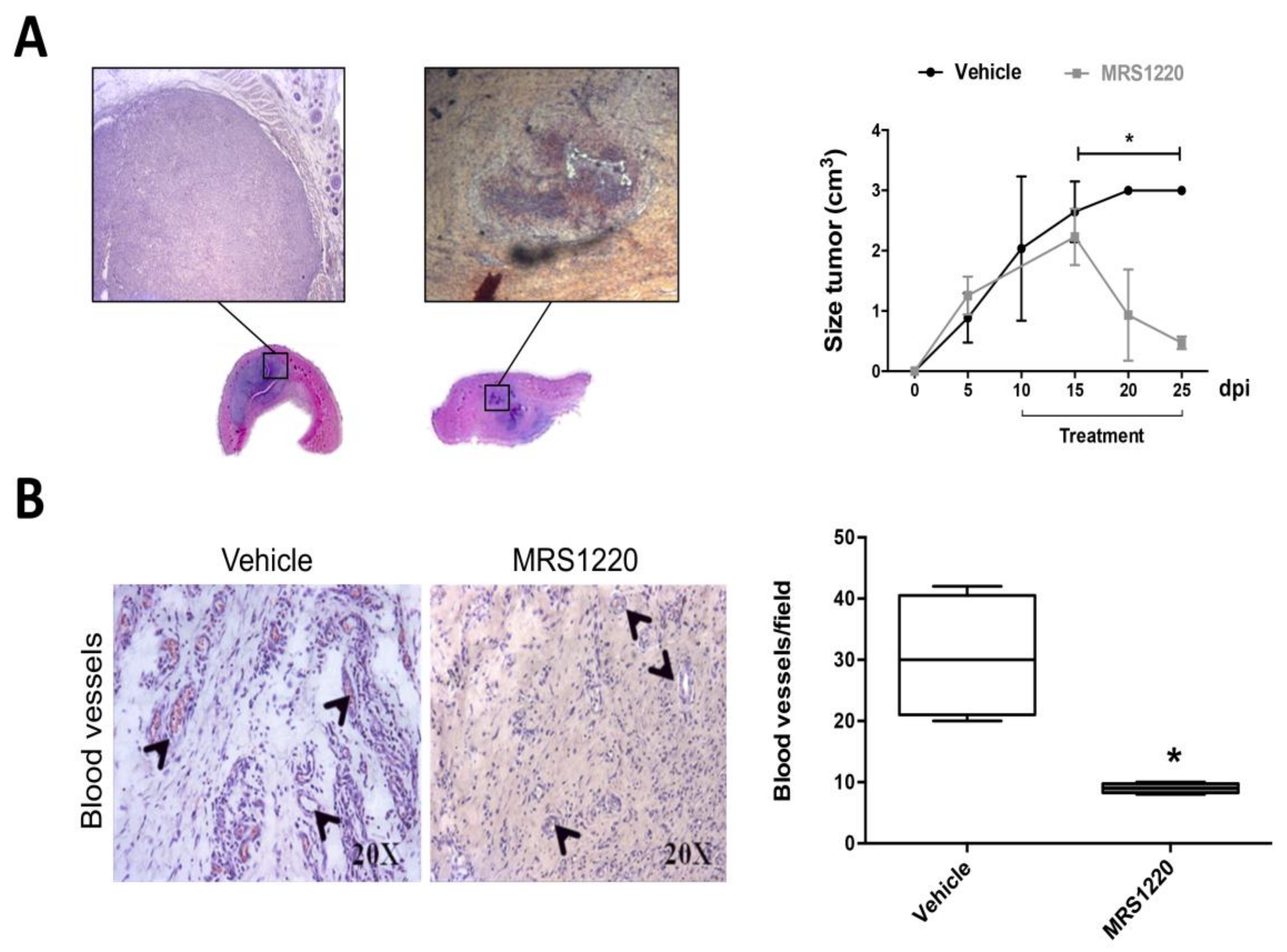
2.4. In Vivo Antagonization of A3AR Decreases Tumor Size and Blood Vessel Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. High Performance Liquid Chromatography (HPLC)
4.3. Western Blot
4.4. Flow Cytometry
4.5. Enzyme-Linked ImmunoSorbent Assay
4.6. In Vivo Studies and Histopathological Analysis
4.7. Statistical Analysis and Artwork
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma Multiforme |
| GSCs | Glioblastoma Stem-Like Cells |
| NSCs | Neural Stem Cells |
| HIF-1α | Hypoxia Inducible Factor 1 |
| VEGF | Vascular Endothelial Growth Factor |
| ECs | Endothelial Cells |
| ARs | Adenosine Receptors |
| A1AR | A1 Adenosine Receptor |
| A2AAR | A2A Adenosine Receptor |
| A2BAR | A2B Adenosine Receptor |
| A3AR | A3 Adenosine Receptor |
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups, National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. Onco. Targets Ther. 2017, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Shaifer, C.A.; Huang, J.; Lin, P.C. Glioblastoma cells incorporate into tumor vasculature and contribute to vascular radioresistance. Int. J. Cancer 2010, 127, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Golden, E.B.; Sivakumar, W.; Wang, W.; Pen, L.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Glioma-associated endothelial cells are chemoresistant to temozolomide. J. Neurooncol. 2009, 95, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasan, R.; Mohanam, I.; Govindarajan, K.; Mohanam, S. Glioma cells suppress hypoxia-induced endothelial cell apoptosis and promote angiogenic process. Int. J. Oncol. 2007, 30, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Coma, S.; Shimizu, A.; Klagsbrun, M. Hypoxia induces tumor and endothelial cell migration in a Semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor Neuropilin 2. Cell Adh. Migr. 2011, 5, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’avella, D.; Basso, G. Intratumoural hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.; Chalmers, A.J. Radioresistance of glioma stem cells: Intrinsic characteristic or property of the ‘microenvironment-stem cell unit’? Mol. Oncol. 2011, 5, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Hitomi, M.; Venere, M.; Flavahan, W.A.; Yang, K.; Kim, Y.; Minhas, S.; Rich, J.N.; Hjelmeland, A.B. Glioma Stem Cell Maintenance: The Role of the Microenvironment. Curr. Pharm. Des. 2011, 17, 2386–2401. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Turcotte, M.; Spring, K.; Pommey, S.; Royal, I.; Stagg, J. Anti-CD73 therapy impairs tumor angiogenesis. Int. J. Cancer 2014, 134, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2α in pulmonary endothelial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine Receptors and Cancer. Biochim. Biophys. Acta 2009, 1808, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Asp. Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Quezada, C.; Garrido, W.; Oyarzún, C.; Fernández, K.; Segura, R.; Melo, R.; Casanello, P.; Sobrevia, L.; San Martín, R. 5′-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J. Cell. Physiol. 2013, 228, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; López, M.N.; Melo, R.; Oyarzún, C.; San Martín, R.; et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Zappelli, E.; Natali, L.; Martini, C.; Trincavelli, M.L. Modulation of A1 and A2B adenosine receptor activity: A new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014, 5, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Tao, L.; Shen, S.; Chen, L. Hypoxia induced CCL28 promotes angiogenesis in lung adenocarcinoma by targeting CCR3 on endothelial cells. Sci. Rep. 2016, 6, 27152. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 2001, 61, 443–448. [Google Scholar] [CrossRef]
- Koszałka, P.; Gołuńska, M.; Urban, A.; Stasiłojć, G.; Stanisławowski, M.; Majewski, M.; Składanowski, A.C.; Bigda, J. Specific Activation of A3, A2A and A1 Adenosine Receptors in CD73-Knockout Mice Affects B16F10 Melanoma Growth, Neovascularization, Angiogenesis and Macrophage Infiltration. PLoS ONE 2016, 11, e151420. [Google Scholar] [CrossRef] [PubMed]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem. Pharmacol. 2006, 72, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.E.; Sharp, T.V.; McKay, T.R. The role of hypoxia in stem cell potency and differentiation. Regen. Med. 2013, 8, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Guo, S.; Yang, L. Effects of all-trans retinoic acid on VEGF and HIF-1α expression in glioma cells under normoxia and hypoxia and its anti-angiogenic effect in an intracerebral glioma model. Mol. Med. Rep. 2014, 10, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rahimpour, S.; Nesvick, C.L.; Zhang, X.; Ma, J.; Zhang, M.; Zhang, G.; Wang, L.; Yang, C.; Hong, C.S.; et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: Implications for bevacizumab antiangiogenic therapy. Oncotarget 2015, 6, 11882–11893. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.Q.; Liu, Y.J.; Ke, Y.Q.; Chen, L.; Jiang, X.D.; Jiang, C.L.; Ye, W. All-trans retinoic acid impairs the vasculogenic mimicry formation ability of U87 stem-like cells through promoting differentiation. Mol. Med. Rep. 2015, 12, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gruslova, A.; Cavazos, D.A.; Miller, J.R.; Breitbart, E.; Cohen, Y.C.; Bangio, L.; Yakov, N.; Soundararajan, A.; Floyd, J.R.; Brenner, A.J. VB-111: A novel anti-vascular therapeutic for glioblastoma multiforme. J. Neurooncol. 2015, 124, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Roa, H.; Gajardo, C.; Troncoso, E.; Fuentealba, V.; Escudero, C.; Yáñez, A.; Sobrevia, L.; Pastor-Anglada, M.; Quezada, C.; San Martin, R. Adenosine mediates transforming growth factor-beta 1 release in kidney glomeruli of diabetic rats. FEBS Lett. 2009, 583, 3192–3198. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. https://doi.org/10.3390/ijms19041228
Rocha R, Torres Á, Ojeda K, Uribe D, Rocha D, Erices J, Niechi I, Ehrenfeld P, San Martín R, Quezada C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. International Journal of Molecular Sciences. 2018; 19(4):1228. https://doi.org/10.3390/ijms19041228
Chicago/Turabian StyleRocha, René, Ángelo Torres, Karina Ojeda, Daniel Uribe, Dellis Rocha, José Erices, Ignacio Niechi, Pamela Ehrenfeld, Rody San Martín, and Claudia Quezada. 2018. "The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia" International Journal of Molecular Sciences 19, no. 4: 1228. https://doi.org/10.3390/ijms19041228