212Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models




Abstract
:1. Introduction
2. Results
2.1. In Vitro Differential Expression of CSPG4 on Human TNBC Cell Lines
2.2. 212Pb-225.28 Reduces the In Vitro Proliferation of Human TNBC Differentiated Cells and CICs
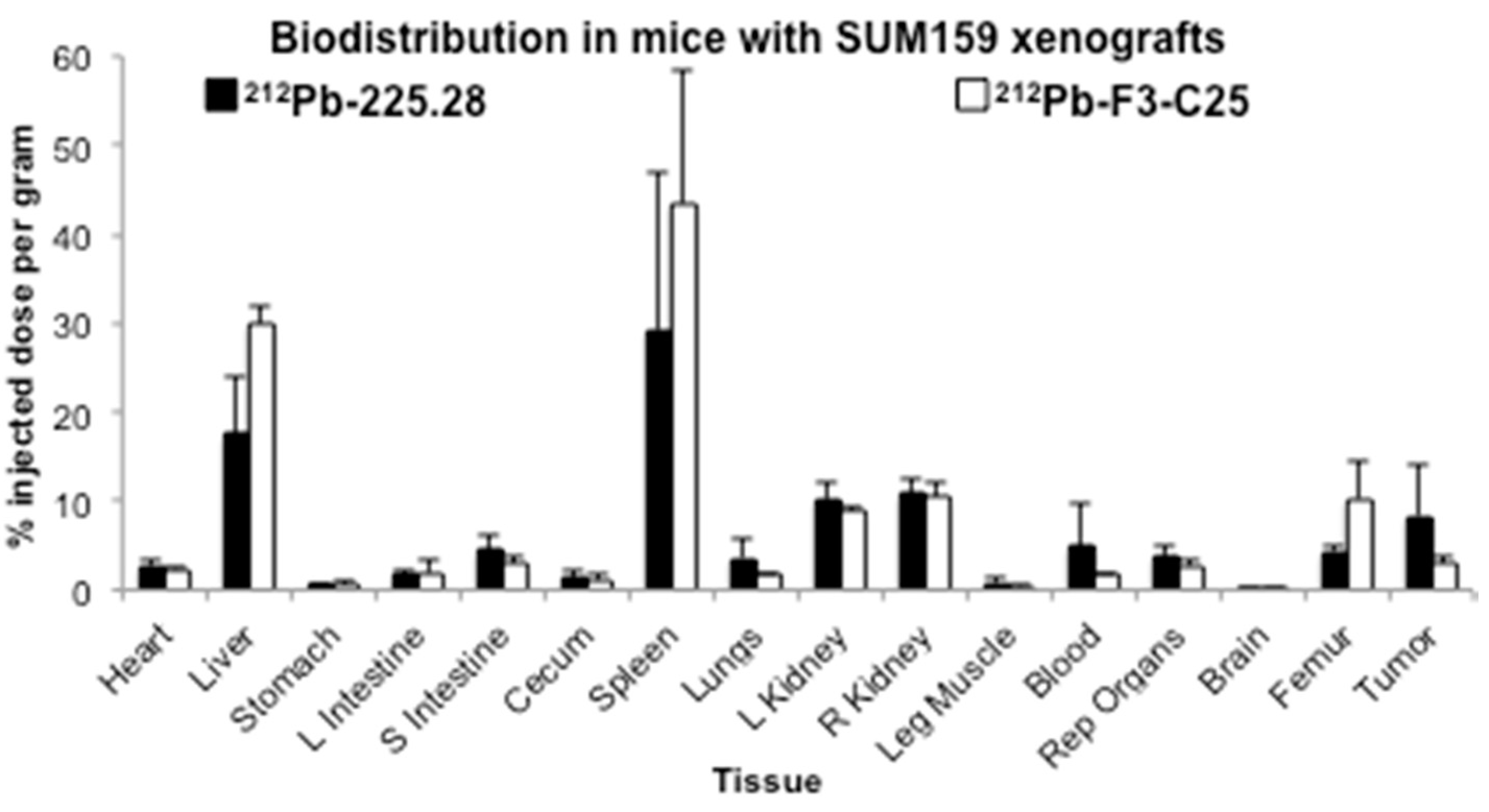
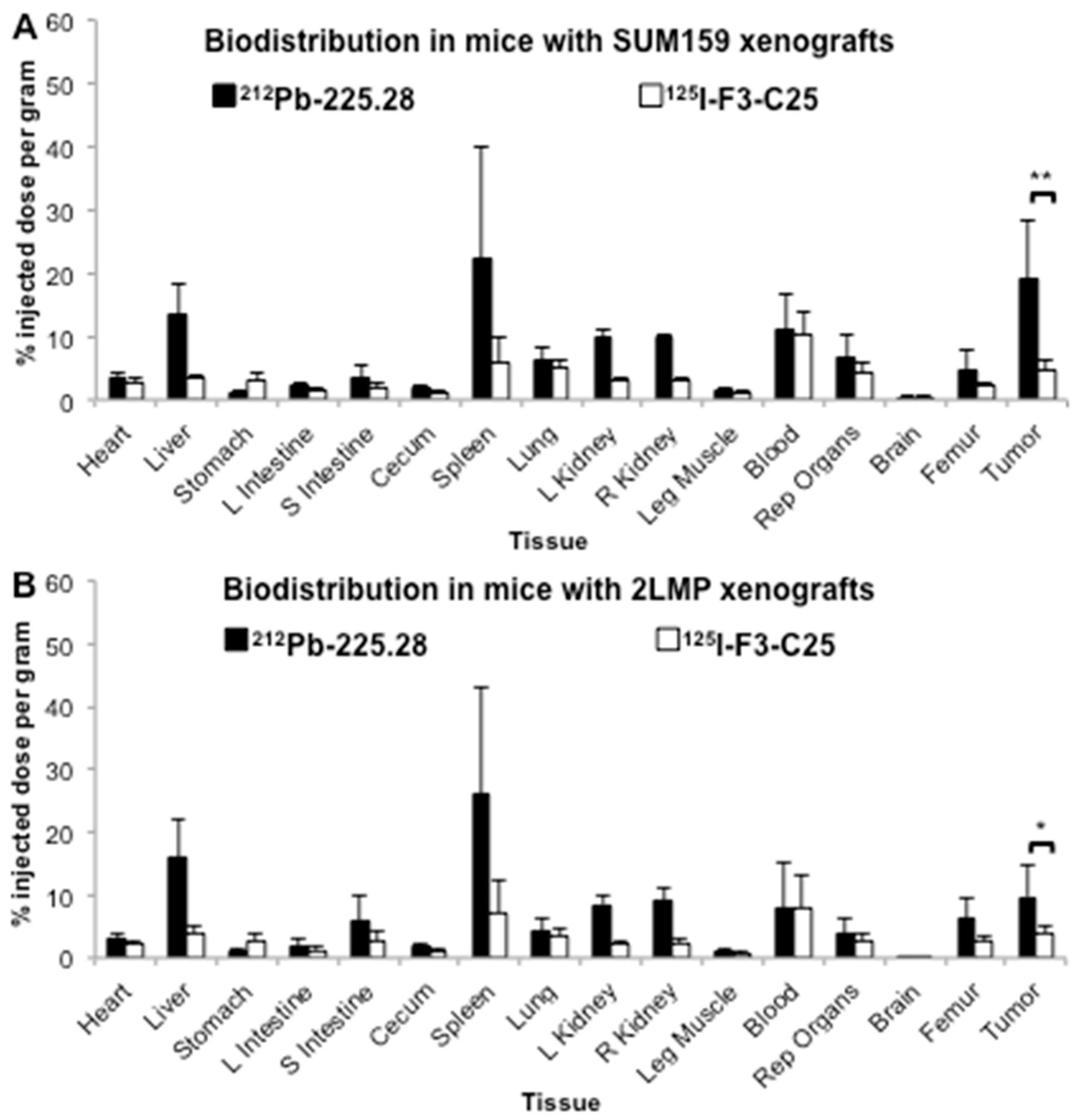
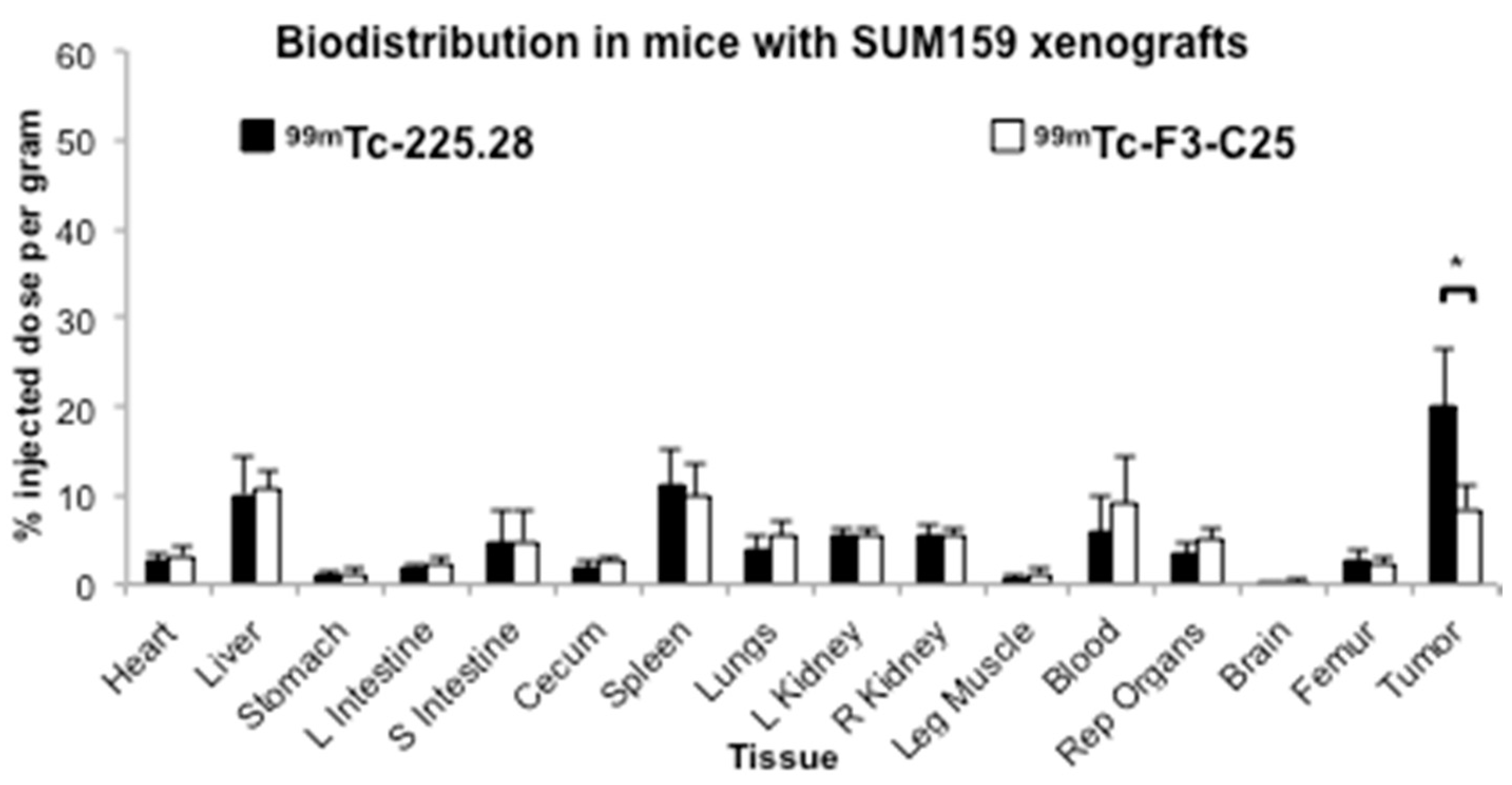
2.3. Tumor Size Influences the Uptake of Radiolabeled mAb 225.28 in Human TNBC Xenografts
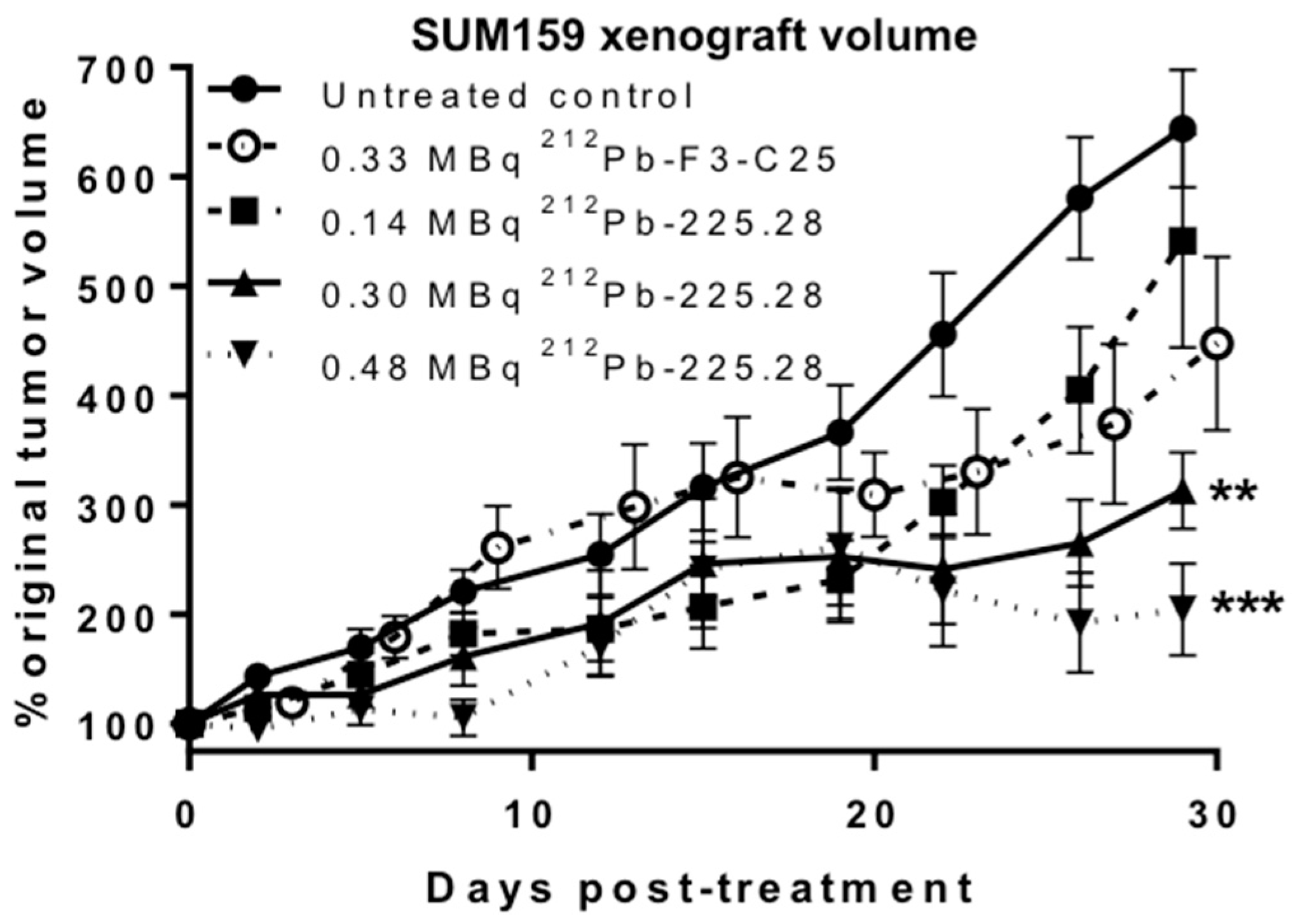
2.4. 212Pb-225.28 Inhibits the Growth of Human TNBC Xenografts
3. Discussion
4. Materials and Methods
4.1. Reagents and Instrumentation
4.2. Radiolabeling
4.3. Human TNBC Cell Lines and CICs
4.4. In Vitro Binding Assays
4.5. In Vitro Clonogenic Survival Assays
4.6. Animal Subjects and Husbandry
4.7. In Vivo Biodistribution and Imaging Studies
4.8. In Vivo Therapy Studies
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TNBC | Triple negative breast cancer |
| CIC | Cancer initiating cell |
| RIT | Radioimmunotherapy |
| CSPG4 | Chondroitin sulfate proteoglycan 4 |
| mAb | Monoclonal antibody |
| RIC | Radioimmunoconjugate |
| HER-2 | Human epidermal growth factor receptor 2 |
| LET | Linear energy transfer |
| keV | Kiloelectron volt |
| i.v. | Intravenous |
| HPGe | High purity germanium |
| TCMC | 2-(4-isothiocyanotobenzyl)-1,4,7,10-tetraaza-1,4,7,10-tetra-(2-carbamoylmethyl)-cyclododecane |
| EDTA | Ethylene diaminetetraacetic acid |
| ITLC | Instant thin layer chromatography |
| ANOVA | Analysis of variance |
| RES | Reticuloendothelial system |
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Telang, N. Putative cancer-initiating stem cells in cell culture models for molecular subtypes of clinical breast cancer. Oncol. Lett. 2015, 10, 3840–3846. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Yu, L.; Sakakura, K.; Visus, C.; Schwab, J.H.; Ferrone, C.R.; Favoino, E.; Koya, Y.; Campoli, M.R.; et al. CSPG4 in cancer: Multiple roles. Curr. Mol. Med. 2010, 10, 419–429. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.K.; Chen, L.; Zhong, W.; Armellino, D.; Yu, J.; Loreth, C.; Follettie, M.; Damelin, M. Breast cancer cells respond differentially to modulation of TGFβ2 signaling after exposure to chemotherapy or hypoxia. Cancer Res. 2015, 75, 4605–4616. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Miragaya, J.; Palafox, M.; Paré, L.; Yoldi, G.; Ferrer, I.; Vila, S.; Galván, P.; Pellegrini, P.; Pérez-Montoyo, H.; Igea, A.; et al. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of a CD49f+ tumor-initiating population. Stem Cell Rep. 2017, 8, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jin, Q.; Wang, X.; Zhu, H.; Ni, Q. Aldehyde dehydrogenase 1 expression is correlated with poor prognosis in breast cancer. Medicine 2017, 96, e7171. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Ma, D.; Lai, L.T.; Simon, J.; Borchardt, P.; Frank, R.K.; Wu, K.; Pellegrini, V.; Curcio, M.J.; Miederer, M.; et al. Tumor therapy with targeted atomic nanogenerators. Science 2001, 294, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Ballangrud, A.M.; Yang, W.H.; Palm, S.; Enmon, R.; Borchardt, P.E.; Pellegrini, V.A.; McDevitt, M.R.; Scheinberg, D.A.; Sgouros, G. Alpha-particle emitting atomic generator (actinium-225)-labeled trastuzumab (herceptin) targeting of breast cancer spheroids: Efficacy versus HER2/neu expression. Clin. Cancer Res. 2004, 10, 4489–4497. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Ma, D.; Abdulla, A.; Brechbiel, M.W. α-particle radioimmunotherapy of disseminated peritoneal disease using a 212Pb-labeled radioimmunoconjugate targeting HER2. Cancer Biother. Radiopharm. 2005, 20, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Elgqvist, J.; Frost, S.; Pouget, J.-P.; Albertsson, P. The potential and hurdles of targeted alpha therapy—Clinical trials and beyond. Front. Oncol. 2014, 3, 324. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hedayati, M.; Hobbs, R.F.; Shao, C.; Bruchertseifer, F.; Morgenstern, A.; DeWeese, T.L.; Sgouros, G. Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-EGFR antibody. Mol. Cancer Ther. 2013, 12, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Aksnes, A.-K.; Naume, B.; Garcia, C.; Jerusalem, G.; Piccart, M.; Vobecky, N.; Thuresson, M.; Flamen, P. A phase IIa, nonrandomized study of radium-223 dichloride in advanced breast cancer patients with bone-dominant disease. Breast Cancer Res. Treat. 2014, 145, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Takalkar, A.; Adams, S.; Subbiah, V. Radium-223 dichloride bone-targeted alpha particle therapy for hormone-refractory breast cancer metastatic to bone. Exp. Hematol. Oncol. 2014, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Akabani, G.; Carlin, S.; Welsh, P.; Zalutsky, M.R. In Vitro cytotoxicity of 211At-labeled trastuzumab in human breast cancer cell lines: Effect of specific activity and HER2 receptor heterogeneity on survival fraction. Nucl. Med. Biol. 2006, 33, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Shahverdi, K.; Huso, D.L.; Esaias, C.; Fox, J.; Liedy, A.; Zhang, Z.; Reilly, R.T.; Apostolidis, C.; Morgenstern, A.; et al. 213Bi (α-emitter)-antibody targeting of breast cancer metastases in the neu-N transgenic mouse model. Cancer Res. 2008, 68, 3873–3880. [Google Scholar] [CrossRef] [PubMed]
- Boskovitz, A.; McLendon, R.E.; Okamura, T.; Sampson, J.H.; Bigner, D.D.; Zalutsky, M.R. Treatment of HER2-positive breast carcinomatous meningitis with intrathecal administration of α-particle-emitting 211At-labeled trastuzumab. Nucl. Med. Biol. 2009, 36, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hobbs, R.F.; Vajravelu, R.; Huso, D.L.; Esaias, C.; Apostolidis, C.; Morgenstern, A.; Sgouros, G. Radioimmunotherapy of breast cancer metastases with α-particle emitter 225Ac: Comparing efficacy with 213Bi and 90Y. Cancer Res. 2009, 69, 8941–8948. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, M.; Song, H.; Thompson, S.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Immunoliposomal delivery of 213Bi for α-emitter targeting of metastatic breast cancer. Cancer Res. 2010, 70, 6815–6823. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Heyerdahl, H.; Bruland, Ø.S.; Borrebæk, J.; Nesland, J.; Dahle, J. Experimental α-particle radioimmunotherapy of breast cancer using 227Th-labeled p-benzyl-DOTA-trastuzumab. EJNMMI Res. 2011, 1, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Jin, K.; Song, H.; Park, S.; Huso, D.L.; Zhang, Z.; Liangfeng, H.; Zhu, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Effective treatment of ductal carcinoma in situ with a HER-2-targeted alpha-particle emitting radionuclide in a preclinical model of human breast cancer. Oncotarget 2016, 7, 33306–33315. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.; Fan, J.; Ferrone, S.; Zinn, K.; Buchsbaum, D. Targeted radioimmunotherapy of triple negative breast cancer with CSPG4-specific 212Pb-labeled monoclonal antibody. J. Nucl. Med. 2016, 57, 114. [Google Scholar]
- Nedrow, J.R.; Josefsson, A.; Park, S.; Bäck, T.; Hobbs, R.F.; Brayton, C.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Pharmacokinetics, microscale distribution, and dosimetry of alpha-emitter-labeled anti-PD-L1 antibodies in an immune competent transgenic breast cancer model. EJNMMI Res. 2017, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sempkowski, M.; Holleran, T.; Linz, T.; Bertalan, T.; Josefsson, A.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Alpha-particle radiotherapy: For large solid tumors diffusion trumps targeting. Biomaterials 2017, 130, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Wang, Y.; Wang, X.; Schwab, J.H.; Ferrone, S.; Ferrone, C.R. Novel tumor antigen-specific monoclonal antibody-based immunotherapy to eradicate both differentiated cancer cells and cancer-initiating cells in solid tumors. Semin. Oncol. 2014, 41, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S.; Imai, K.; Natali, P.G.; Ferrone, S. Distribution and molecular characterization of a cell-surface and a cytoplasmic antigen detectable in human melanoma cells with monoclonal antibodies. Int. J. Cancer 1981, 28, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Horak, E.; Hartmann, F.; Garmestani, K.; Wu, C.; Brechbiel, M.; Gansow, O.A. Radioimmunotherapy targeting of HER2/neu oncoprotein on ovarian tumor using lead-212-DOTA-AE1. J. Nucl. Med. 1997, 38, 1944–1950. [Google Scholar] [PubMed]
- Bäck, T.; Chouin, N.; Lindegren, S.; Kahu, H.; Jensen, H.; Albertsson, P.; Palm, S. Cure of human ovarian carcinoma solid xenografts by fractionated α-radioimmunotherapy with 211At-MX35-F(ab′)2: Influence of absorbed tumor dose and effect on long-term survival. J. Nucl. Med. 2017, 58, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Gangrade, A.; Kim, H.; Fan, J.; Ferrone, S.; Ferrone, C.R.; Zinn, K.R.; Buchsbaum, D.J. 212Pb-labeled B7-H3-targeting antibody for pancreatic cancer therapy in mouse models. Nucl. Med. Biol. 2017, 58, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sabbatino, F.; Yu, L.; Favoino, E.; Wang, X.; Ligorio, M.; Ferrone, S.; Schwab, J.H.; Ferrone, C.R. Tumor antigen-specific monoclonal antibody-based immunotherapy, cancer initiating cells and disease recurrence. In Resistance to Immunotherapeutic Antibodies in Cancer; Bonavida, B., Ed.; Springer: New York, NY, USA, 2013; pp. 25–47. [Google Scholar]
- Sgouros, G.; Song, H. Cancer stem cell targeting using the alpha-particle emitter, 213Bi: Mathematical modeling and feasibility analysis. Cancer Biother. Radiopharm. 2008, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Macey, D.J.; Grant, E.J.; Kasi, L.; Rosenblum, M.G.; Zhang, H.Z.; Katz, R.L.; Rieger, P.T.; LeBherz, D.; South, M.; Greiner, J.W.; et al. Effect of recombinant alpha-interferon on pharmacokinetics, biodistribution, toxicity, and efficacy of 131I-labeled monoclonal antibody CC49 in breast cancer: A phase II trial. Clin. Cancer Res. 1997, 3, 1547–1555. [Google Scholar] [PubMed]
- Cornelissen, B.; McLarty, K.; Kersemans, V.; Reilly, R.M. The level of insulin growth factor-1 receptor expression is directly correlated with the tumor uptake of 111In-IGF-1(E3R) in vivo and the clonogenic survival of breast cancer cells exposed in vitro to trastuzumab (Herceptin). Nucl. Med. Biol. 2008, 35, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, D.J. Experimental approaches to increase radiolabeled antibody localization in tumors. Cancer Res. 1995, 55, 5729s–5732s. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.F.; Khazaeli, M.B.; Macey, D.J.; Grizzle, W.E.; Mayo, M.; Schlom, J.; Russell, C.D.; LoBuglio, A.F. Phase II Study of Interferon-enhanced 131I-Labeled High Affinity CC49 Monoclonal Antibody Therapy in Patients with Metastatic Prostate Cancer. Clin. Cancer Res. 1999, 5, 3254s–3258s. [Google Scholar] [PubMed]
- Miao, Y.; Hylarides, M.; Fisher, D.R.; Shelton, T.; Moore, H.; Wester, D.W.; Fritzberg, A.R.; Winkelmann, C.T.; Hoffman, T.; Quinn, T.P. Melanoma therapy via peptide-targeted α-radiation. Clin. Cancer Res. 2005, 11, 5616–5621. [Google Scholar] [CrossRef] [PubMed]
- Su, F.M.; Beaumier, P.; Axworthy, D.; Atcher, R.; Fritzberg, A. Pretargeted radioimmunotherapy in tumored mice using an in vivo 212Pb/212Bi generator. Nucl. Med. Biol. 2005, 32, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Kukis, D.L.; DeNardo, G.L.; DeNardo, S.J.; Mirick, G.R.; Miers, L.A.; Greiner, D.P.; Meares, C.F. Effect of the extent of chelate substitution on the immunoreactivity and biodistribution of 2IT-BAT-Lym-1 immunoconjugates. Cancer Res. 1995, 55, 878–884. [Google Scholar] [PubMed]
- Al-Ejeh, F.; Darby, J.M.; Thierry, B.; Brown, M.P. A simplified suite of methods to evaluate chelator conjugation of antibodies: Effects on hydrodynamic radius and biodistribution. Nucl. Med. Biol. 2009, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Fawwaz, R.A.; Wang, T.S.T.; Estabrook, A.; Rosen, J.M.; Hardy, M.A.; Alderson, P.O.; Srivastava, S.C.; Richards, P.; Ferrone, S. Immunoreactivity and biodistribution of indium-111-labeled monoclonal antibody to a human high molecular weight-melanoma associated antigen. J. Nucl. Med. 1985, 26, 488–492. [Google Scholar] [PubMed]
- Buraggi, G.; Turrin, A.; Cascinelli, N.; Attili, A.; Gasparini, M.; Callegaro, L.; Ferrone, S.; Seregni, E.; Bombardieri, E.; Belli, F. Radioimmunodetection of melanoma: Preliminary results of a prospective study. Int. J. Biol. Markers 1986, 1, 47–54. [Google Scholar] [PubMed]
- Siccardi, A.G.; Buraggi, G.L.; Callegaro, L.; Mariani, G.; Natali, P.G.; Abbati, A.; Bestagno, M.; Caputo, V.; Mansi, L.; Masi, R.; et al. Multicenter study of immunoscintigraphy with radiolabeled monoclonal antibodies in patients with melanoma. Cancer Res. 1986, 46, 4817–4822. [Google Scholar] [PubMed]
- Kasten, B.B.; Arend, R.C.; Katre, A.A.; Kim, H.; Fan, J.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. B7-H3-targeted 212Pb radioimmunotherapy of ovarian cancer in preclinical models. Nucl. Med. Biol. 2017, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Heyerdahl, H.; Abbas, N.; Brevik, E.M.; Mollatt, C.; Dahle, J. Fractionated therapy of HER2-expressing breast and ovarian cancer xenografts in mice with targeted alpha emitting 227Th-DOTA-p-benzyl-trastuzumab. PLoS ONE 2012, 7, e42345. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410. [Google Scholar] [CrossRef] [PubMed]
- Baidoo, K.E.; Milenic, D.E.; Brechbiel, M.W. Methodology for labeling proteins and peptides with lead-212 (212Pb). Nucl. Med. Biol. 2013, 40, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Ferrone, S. Syngeneic anti-idiotypic antisera to murine anti-HLA Class II monoclonal antibodies. J. Immunol. 1987, 139, 1232–1239. [Google Scholar] [PubMed]
- Chappell, L.L.; Dadachova, E.; Milenic, D.E.; Garmestani, K.; Wu, C.; Brechbiel, M.W. Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl. Med. Biol. 2000, 27, 93–100. [Google Scholar] [CrossRef]
- Dadachova, E.; Chappell, L.L.; Brechbiel, M.W. Spectrophotometric method for determination of bifunctional macrocyclic ligands in macrocyclic ligand-protein conjugates. Nucl. Med. Biol. 1999, 26, 977–982. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Kim, H.; Zhai, G.; Liu, Z.; Samuel, S.; Shah, N.; Helman, E.E.; Knowles, J.A.; Stockard, C.R.; Fineberg, N.S.; Grizzle, W.E.; et al. EMMPRIN as a novel target for pancreatic cancer therapy. Anticancer Drugs 2011, 22, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Oliver, P.G.; LoBuglio, A.F.; Zhou, T.; Forero, A.; Kim, H.; Zinn, K.R.; Zhai, G.; Li, Y.; Lee, C.H.; Buchsbaum, D.J. Effect of anti-DR5 and chemotherapy on basal-like breast cancer. Breast Cancer Res. Treat. 2012, 133, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.N.; Maurmann, L.; Mishur, R.J.; Yasui, L.; Gupta, S.; Grayburn, W.S.; Hofstetter, H.; Salley, T. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum–phosphato complexes towards human ovarian cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18314–18319. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aKd ± S.E.M. (nmol/L) | a Binding Sites/Cell ± SEM (×103) | Internalization (%) | |
|---|---|---|---|
| Adherent SUM159 | 0.6 ± 0.1 | 29 ± 4.2 | 56 |
| CIC SUM159 | 0.3 ± 0.1 | 49 ± 12 | 57 |
| Adherent 2LMP | 0.5 ± 0.1 | b 3.4 | 49 |
| CIC 2LMP | 0.5 ± 0.2 | b 7.7 | 47 |
| Cells | a IC50 ± SEM (kBq/mL) | |
|---|---|---|
| 212Pb-225.28 | 212Pb-F3-C25 | |
| Adherent SUM159 | 22 ± 10 | 144 ± 19 |
| CIC SUM159 | 12 ± 3 | 86 ± 10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasten, B.B.; Oliver, P.G.; Kim, H.; Fan, J.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. 212Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models. Int. J. Mol. Sci. 2018, 19, 925. https://doi.org/10.3390/ijms19040925
Kasten BB, Oliver PG, Kim H, Fan J, Ferrone S, Zinn KR, Buchsbaum DJ. 212Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models. International Journal of Molecular Sciences. 2018; 19(4):925. https://doi.org/10.3390/ijms19040925
Chicago/Turabian StyleKasten, Benjamin B., Patsy G. Oliver, Harrison Kim, Jinda Fan, Soldano Ferrone, Kurt R. Zinn, and Donald J. Buchsbaum. 2018. "212Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models" International Journal of Molecular Sciences 19, no. 4: 925. https://doi.org/10.3390/ijms19040925
APA StyleKasten, B. B., Oliver, P. G., Kim, H., Fan, J., Ferrone, S., Zinn, K. R., & Buchsbaum, D. J. (2018). 212Pb-Labeled Antibody 225.28 Targeted to Chondroitin Sulfate Proteoglycan 4 for Triple-Negative Breast Cancer Therapy in Mouse Models. International Journal of Molecular Sciences, 19(4), 925. https://doi.org/10.3390/ijms19040925