Baicalein Rescues Delayed Cooling via Preservation of Akt Activation and Akt-Mediated Phospholamban Phosphorylation

Abstract
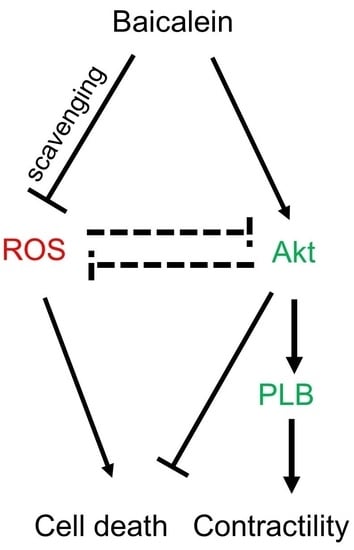
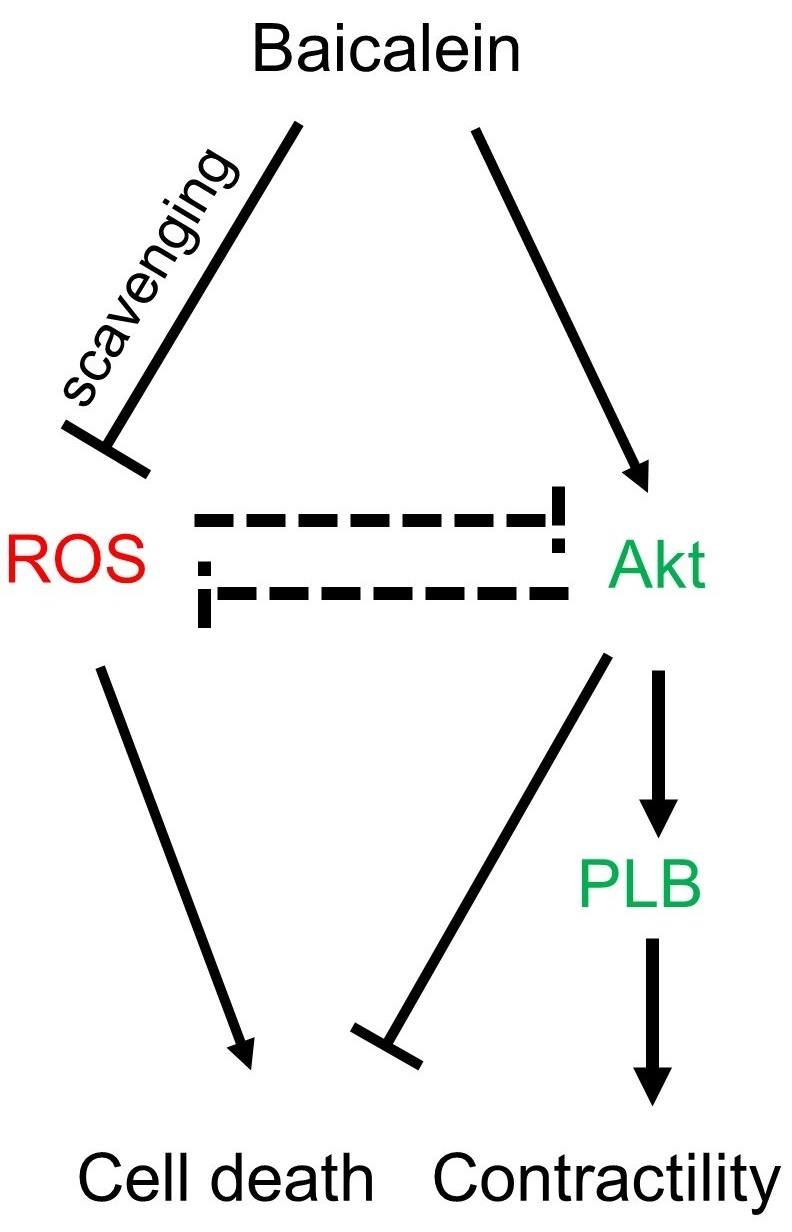
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
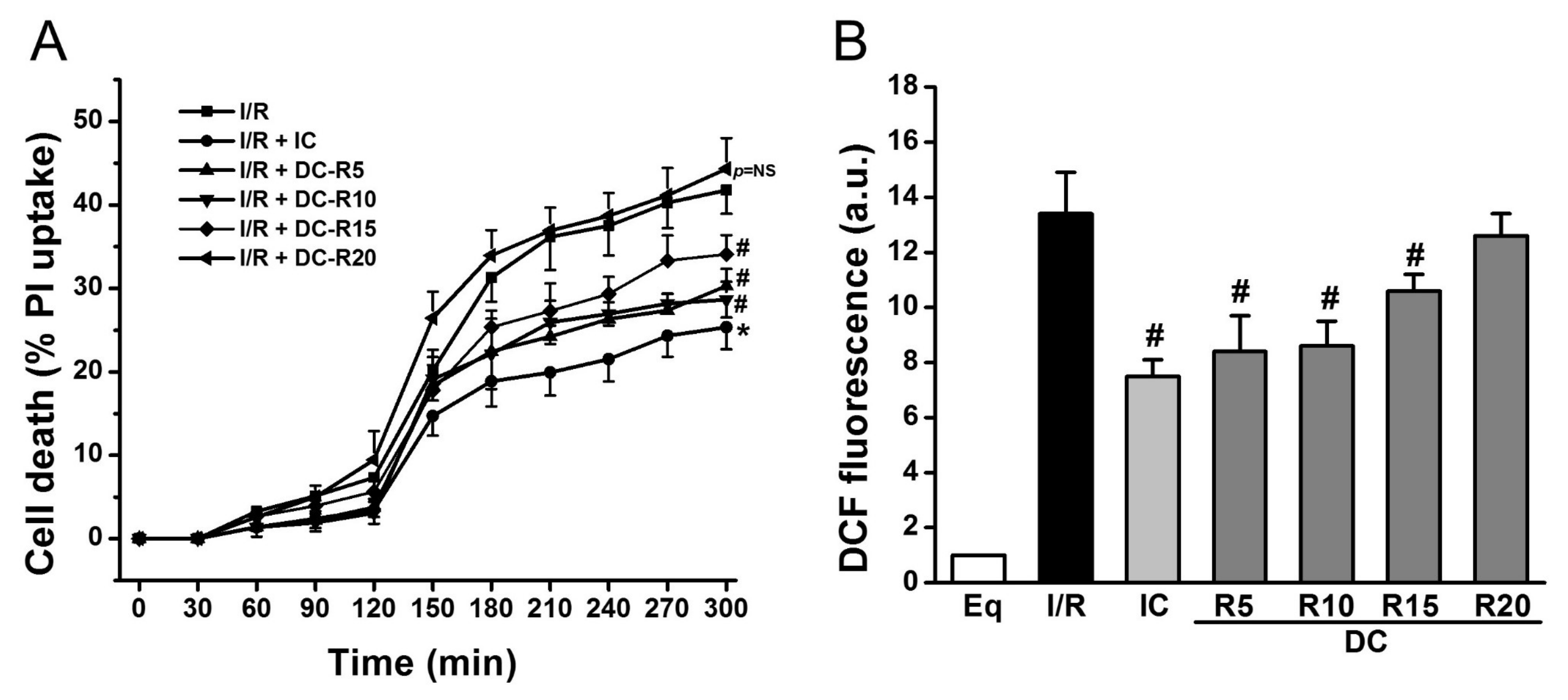
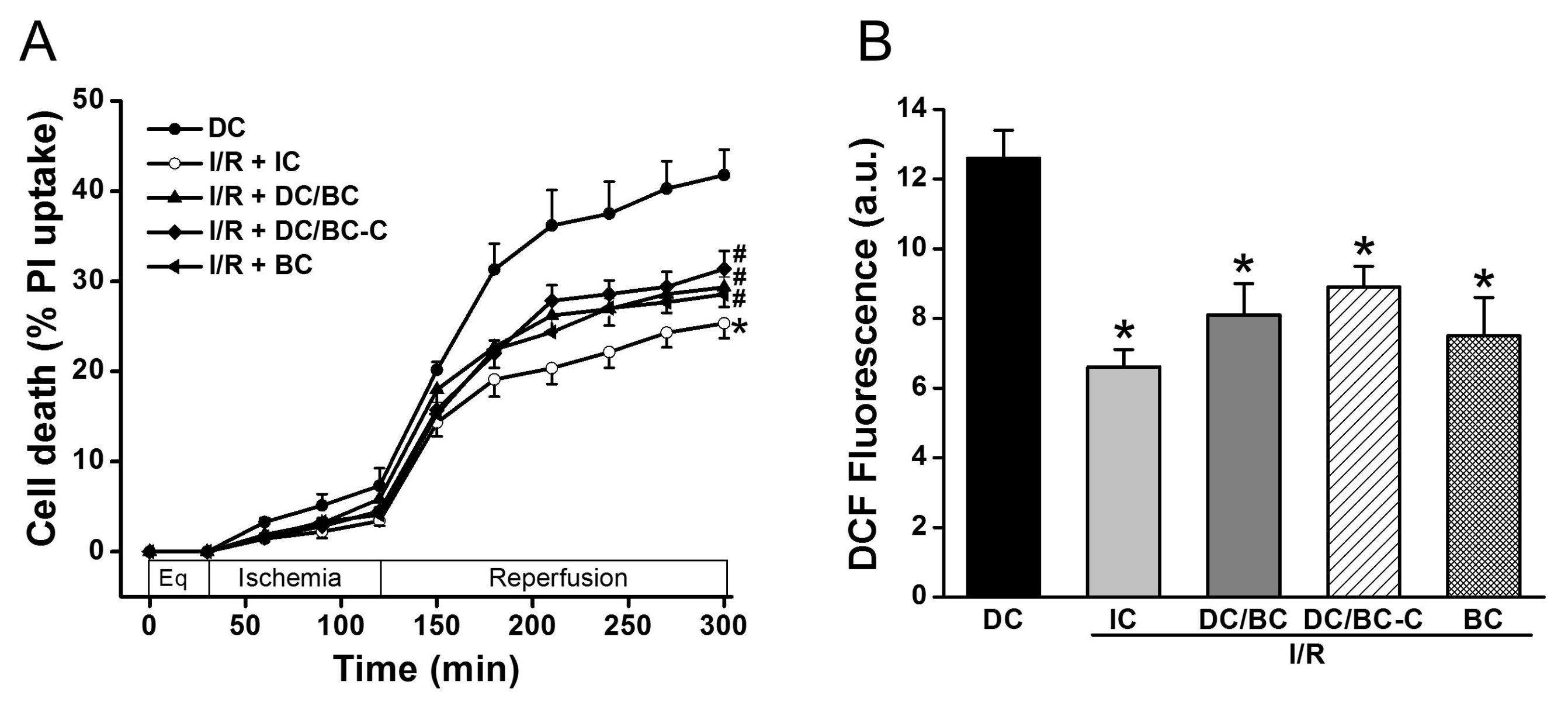
2.1. Delayed Cooling-Diminished Effects of Intraischemic Cooling on Cell Death and ROS Generation
2.2. Baicalein Rescued the Delayed Cooling When Administered at Both the Start of Reperfusion and the Initiation of Cooling during Reperfusion
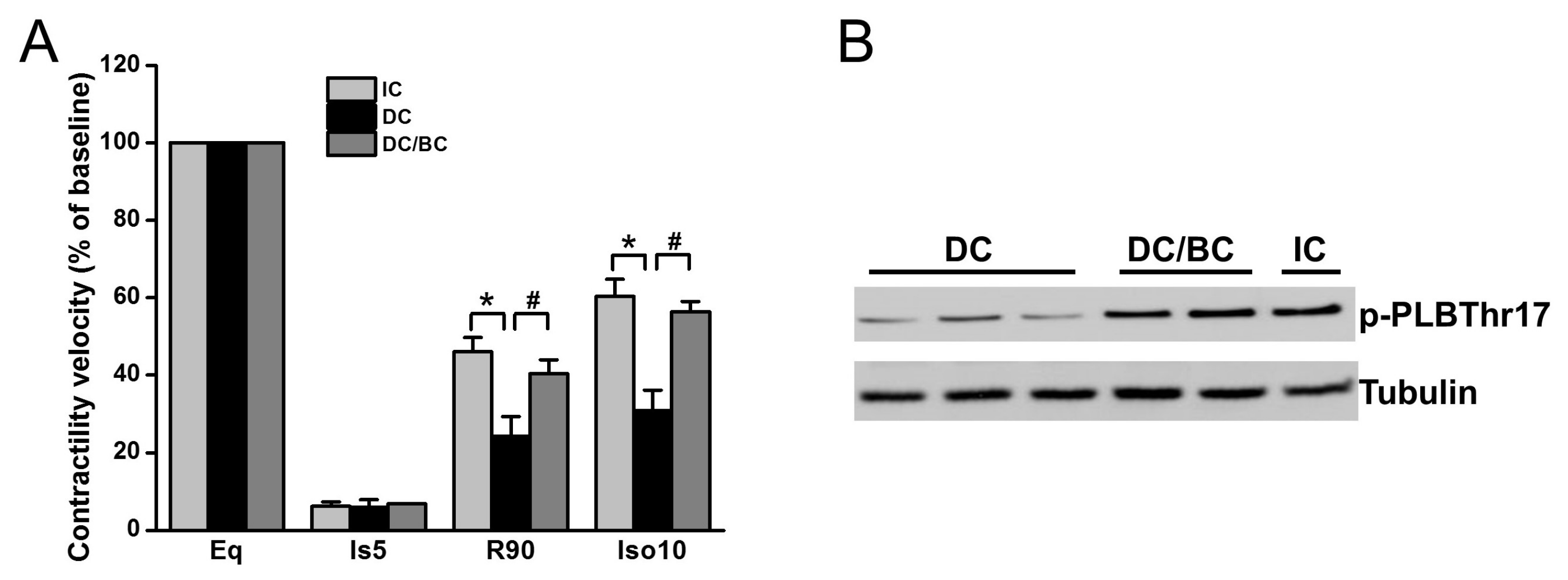
2.3. Baicalein Enhanced Contractility Recovery and Phospholamban Phosphorylation
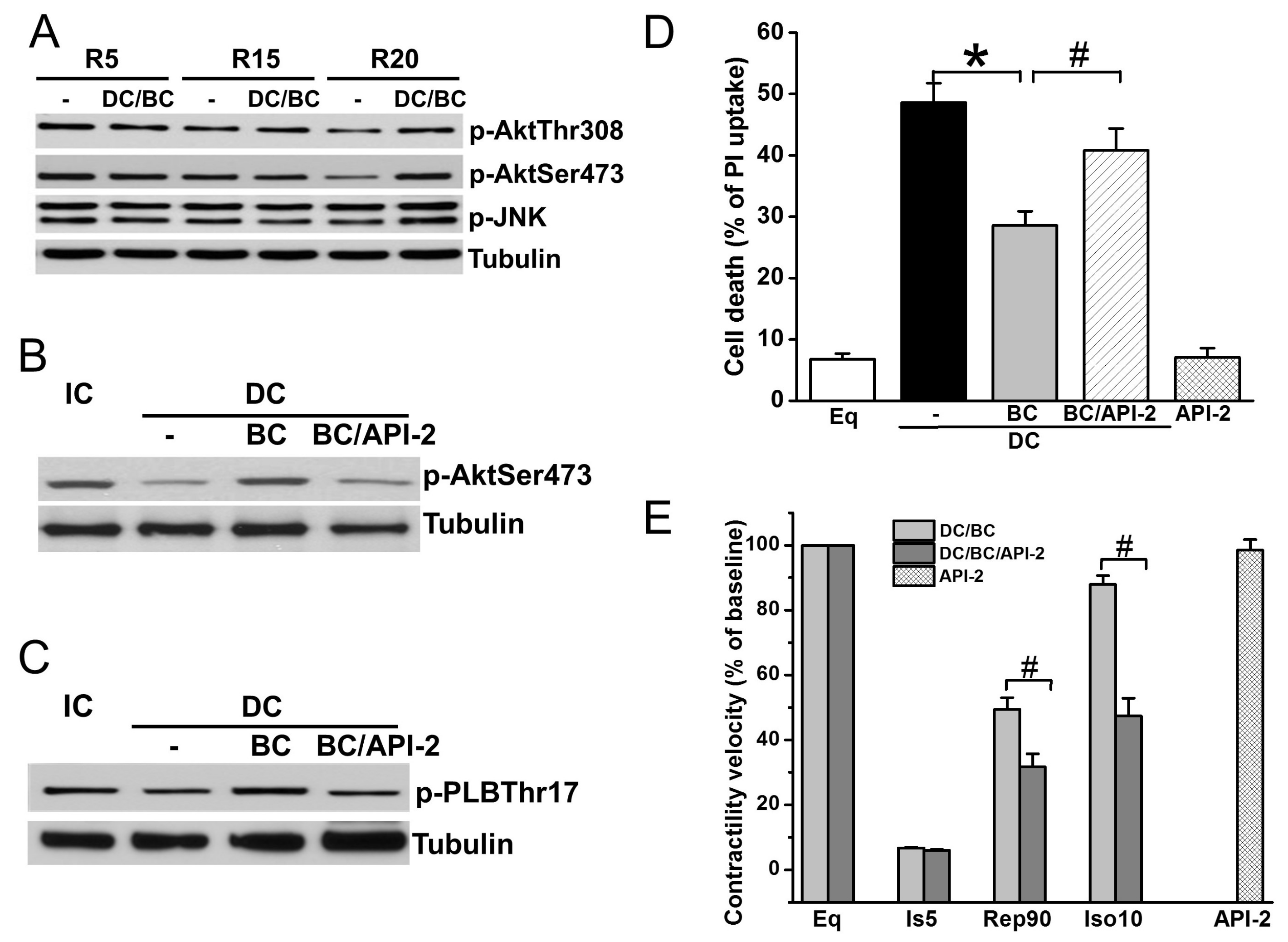
2.4. Baicalein Protection in the Context of Delayed Cooling Was Mediated by Akt Phosphorylation
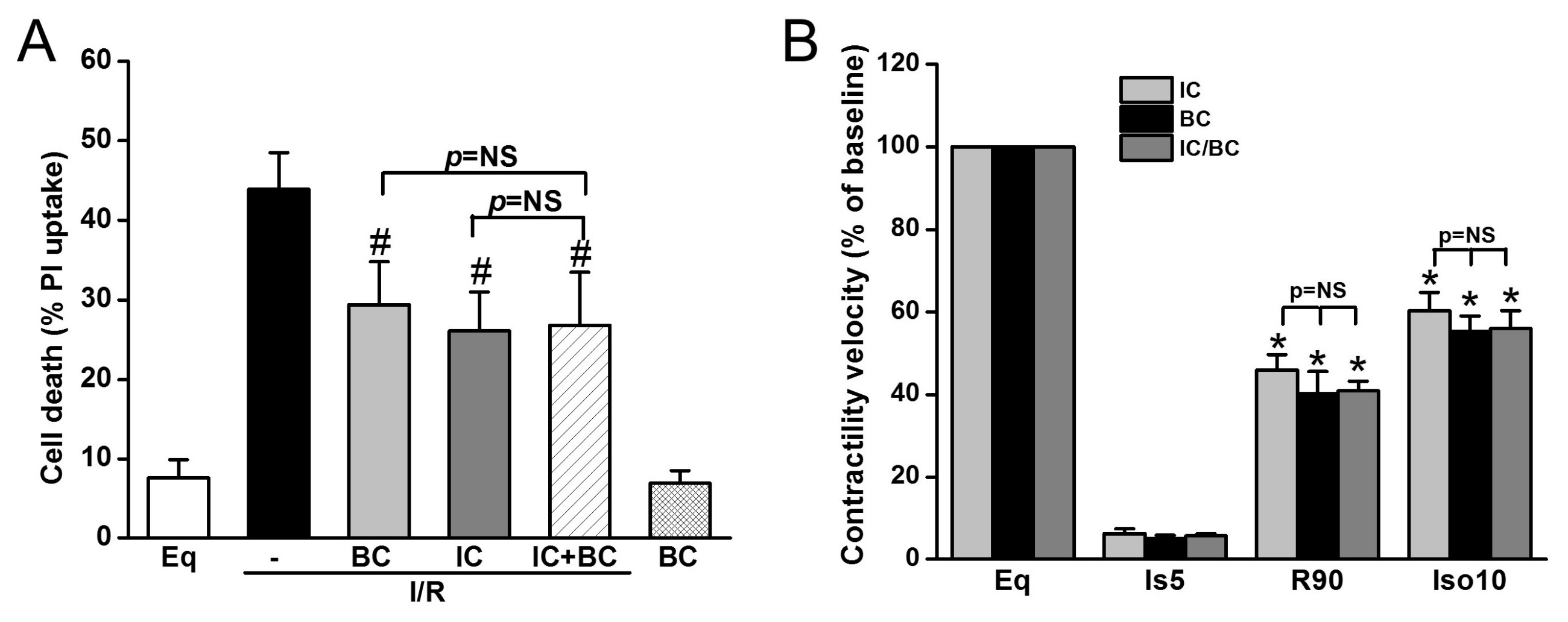
2.5. Combined Treatments of Baicalein and Intraischemic Cooling Exerts No Synergistic Effects
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mouse Cardiomyocyte Culture
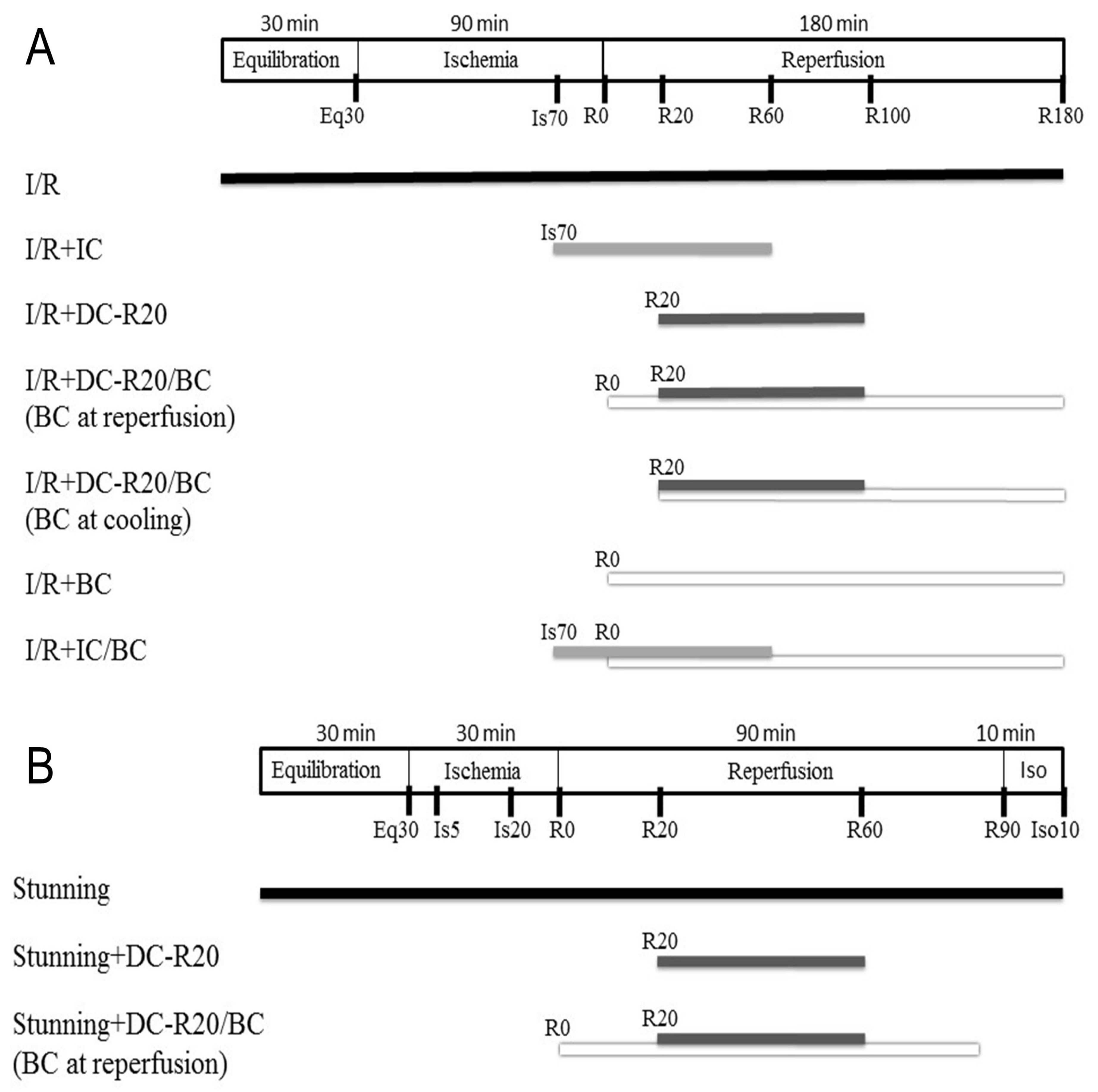
4.3. Cooling Protocol in I/R and Stunning Models
4.4. Cell Viability
4.5. Hypoxia Box
4.6. Measurement of Intracellular ROS
4.7. Western Blot Analysis
4.8. Measurement of the Contractile Velocity
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jennings, R.B.; Murry, C.E.; Steenbergen, C., Jr.; Reimer, K.A. Development of cell injury in sustained acute ischemia. Circulation 1990, 82, II2–12. [Google Scholar] [PubMed]
- Bolli, R.; Patel, B.S.; Zhu, W.X.; O’Neill, P.G.; Hartley, C.J.; Charlat, M.L.; Roberts, R. The iron chelator desferrioxamine attenuates postischemic ventricular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 1987, 253, H1372–H1380. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.E.; Vercellotti, G.M.; Jacob, H.S.; Pieper, G.M.; Gross, G.J. Evidence for a role of iron-catalyzed oxidants in functional and metabolic stunning in the canine heart. Circ. Res. 1988, 63, 351–360. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.L.; Arrell, D.K.; Van Eyk, J.E. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ. Res. 1999, 84, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.D.; Atar, D.; Liu, Y.; Perez, N.G.; Murphy, A.M.; Marban, E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ. Res. 1997, 80, 393–399. [Google Scholar] [PubMed]
- Li, J.; Wang, H.; Zhong, Q.; Zhu, X.; Chen, S.J.; Qian, Y.; Costakis, J.; Bunney, G.; Beiser, D.G.; Leff, A.R.; et al. A novel pharmacological strategy by PTEN inhibition for improving metabolic resuscitation and survival after mouse cardiac arrest. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1414–H1422. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar] [PubMed]
- Hale, S.L.; Kloner, R.A. Mild hypothermia as a cardioprotective approach for acute myocardial infarction: Laboratory to clinical application. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kanemoto, S.; Matsubara, M.; Noma, M.; Leshnower, B.G.; Parish, L.M.; Jackson, B.M.; Hinmon, R.; Hamamoto, H.; Gorman, J.H., 3rd; Gorman, R.C. Mild hypothermia to limit myocardial ischemia-reperfusion injury: Importance of timing. Ann. Thorac. Surg. 2009, 87, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.H.; Chang, W.T.; Chan, K.C.; Wojcik, K.R.; Hsu, C.W.; Li, C.Q.; Li, J.; Anderson, T.; Qin, Y.; Becker, L.B.; et al. Hypothermia-induced cardioprotection using extended ischemia and early reperfusion cooling. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1995–H2003. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.H.; Sharp, W.W.; Wojcik, K.R.; Li, C.Q.; Han, M.; Chang, W.T.; Ramachandran, S.; Li, J.; Hamann, K.J.; Vanden Hoek, T.L. Therapeutic hypothermia cardioprotection via Akt- and nitric oxide-mediated attenuation of mitochondrial oxidants. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2164–H2173. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N. Engl. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Huang, K.; Yang, X.; Xu, H. Free radical scavenging and antioxidant activities of flavonoids extracted from the radix of Scutellaria baicalensis Georgi. Biochim. Biophys. Acta 1999, 1472, 643–650. [Google Scholar] [CrossRef]
- Shao, Z.H.; Li, C.Q.; Vanden Hoek, T.L.; Becker, L.B.; Schumacker, P.T.; Wu, J.A.; Attele, A.S.; Yuan, C.S. Extract from Scutellaria baicalensis Georgi attenuates oxidant stress in cardiomyocytes. J. Mol. Cell. Cardiol. 1999, 31, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Shao, Z.H.; Yin, J.J.; Mehendale, S.; Wang, C.Z.; Qin, Y.; Li, J.; Chen, W.J.; Chien, C.T.; Becker, L.B.; et al. Comparative effects of flavonoids on oxidant scavenging and ischemia-reperfusion injury in cardiomyocytes. Eur. J. Pharmacol. 2007, 566, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.H.; Vanden Hoek, T.L.; Qin, Y.; Becker, L.B.; Schumacker, P.T.; Li, C.Q.; Dey, L.; Barth, E.; Halpern, H.; Rosen, G.M.; et al. Baicalein attenuates oxidant stress in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H999–H1006. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Shao, Z.H.; Li, C.Q.; Vanden Hoek, T.L.; Li, J. Baicalein protects cardiomyocytes against mitochondrial oxidant injury associated with JNK inhibition and mitochondrial Akt activation. Am. J. Chin. Med. 2014, 42, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Liu, G.S.; Cohen, M.V.; Downey, J.M. Mild hypothermia reduces infarct size in the beating rabbit heart: A practical intervention for acute myocardial infarction? Basic Res. Cardiol. 1998, 93, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Dae, M.W.; Gao, D.W.; Sessler, D.I.; Chair, K.; Stillson, C.A. Effect of endovascular cooling on myocardial temperature, infarct size, and cardiac output in human-sized pigs. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1584–H1591. [Google Scholar] [CrossRef] [PubMed]
- Chenoune, M.; Lidouren, F.; Adam, C.; Pons, S.; Darbera, L.; Bruneval, P.; Ghaleh, B.; Zini, R.; Dubois-Rande, J.L.; Carli, P.; et al. Ultrafast and whole-body cooling with total liquid ventilation induces favorable neurological and cardiac outcomes after cardiac arrest in rabbits. Circulation 2011, 124, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piktel, J.S.; Rosenbaum, D.S.; Wilson, L.D. Mild hypothermia decreases arrhythmia susceptibility in a canine model of global myocardial ischemia. Crit. Care Med. 2012, 40, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fettiplace, M.; Chen, S.J.; Steinhorn, B.; Shao, Z.; Zhu, X.; Li, C.; Harty, S.; Weinberg, G.; Vanden Hoek, T.L. Lipid emulsion rapidly restores contractility in stunned mouse cardiomyocytes: A comparison with therapeutic hypothermia. Crit. Care Med. 2014, 42, e734–e740. [Google Scholar] [CrossRef] [PubMed]
- Catalucci, D.; Latronico, M.V.; Ceci, M.; Rusconi, F.; Young, H.S.; Gallo, P.; Santonastasi, M.; Bellacosa, A.; Brown, J.H.; Condorelli, G. Akt increases sarcoplasmic reticulum Ca2+ cycling by direct phosphorylation of phospholamban at Thr17. J. Biol. Chem. 2009, 284, 28180–28187. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Li, J.; Haung, H.H.; Liu, H.; Han, M.; Ramachandran, S.; Li, C.Q.; Sharp, W.W.; Hamann, K.J.; Yuan, C.S.; et al. Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J. Cell. Biochem. 2011, 112, 2873–2881. [Google Scholar] [CrossRef] [PubMed]
- Vanden Hoek, T.L.; Li, C.; Shao, Z.; Schumacker, P.T.; Becker, L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997, 29, 2571–2583. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Prado, R.; Toledo-Pereyra, L.H. The molecular events underlying ischemia/reperfusion injury. Transpl. Proc. 2002, 34, 2518–2519. [Google Scholar] [CrossRef]
- Mikkelsen, M.E.; Christie, J.D.; Abella, B.S.; Kerlin, M.P.; Fuchs, B.D.; Schweickert, W.D.; Berg, R.A.; Mosesso, V.N.; Shofer, F.S.; Gaieski, D.F. Use of therapeutic hypothermia after in-hospital cardiac arrest. Crit. Care Med. 2013, 41, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Zhao, H.; Maier, C.M.; Steinberg, G.K. The protective effect of early hypothermia on PTEN phosphorylation correlates with free radical inhibition in rat stroke. J. Cereb. Blood Flow Metabol. 2009, 29, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Wei, Y.; Guo, M. Iron-binding and anti-Fenton properties of baicalein and baicalin. J. Inorg. Biochem. 2009, 103, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chang, W.T.; Li, C.Q.; Lee, C.; Huang, H.H.; Hsu, C.W.; Chen, W.J.; Zhu, X.; Wang, C.Z.; Vanden Hoek, T.L.; et al. Baicalein Preventive Treatment Confers Optimal Cardioprotection by PTEN/Akt/NO Activation. Am. J. Chin. Med. 2017, 45, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Xu, K.; Cai, F.; Gu, J.; Ma, L.; Chen, J. Neuroprotection by baicalein in ischemic brain injury involves PTEN/AKT pathway. J. Neurochem. 2010, 112, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Waite, K.A.; Eng, C. Protean PTEN: Form and function. Am. J. Hum. Genet. 2002, 70, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, M.; Rovini, A.; Bourgarel-Rey, V.; Honore, S.; Bastonero, S.; Braguer, D.; Carre, M. ROS-mediated EB1 phosphorylation through Akt/GSK3β pathway: Implication in cancer cell response to microtubule-targeting agents. Oncotarget 2014, 5, 3408–3423. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Wu, Y.; Lu, X.; Chidiac, P.; Wang, G.; Feng, Q. Maternal diabetes up-regulates NOX2 and enhances myocardial ischaemia/reperfusion injury in adult offspring. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.H. Future targets and cascades for neuroprotective strategies. Stroke 2004, 35, 2748–2750. [Google Scholar] [CrossRef] [PubMed]
- Lekli, I.; Mukherjee, S.; Ray, D.; Gurusamy, N.; Kim, Y.H.; Tosaki, A.; Engelman, R.M.; Ho, Y.S.; Das, D.K. Functional recovery of diabetic mouse hearts by glutaredoxin-1 gene therapy: Role of Akt-FoxO-signaling network. Gene Ther. 2010, 17, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Hao, Z.; Zhang, S.; Wei, M.; Lu, B.; Wang, Z.; Ji, L. Baicalein and baicalin alleviate acetaminophen-induced liver injury by activating Nrf2 antioxidative pathway: The involvement of ERK1/2 and PKC. Biochem. Pharmacol. 2018, 150, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.J.; Chen, Y.S.; Huang, H.S.; Ma, M.C. Hypoxic preconditioning protects rat hearts against ischemia-reperfusion injury via the arachidonate12-lipoxygenase/transient receptor potential vanilloid 1 pathway. Basic Res. Cardiol. 2014, 109, 414. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.B.; Lee, C.S. Baicalein attenuates proteasome inhibition-induced apoptosis by suppressing the activation of the mitochondrial pathway and the caspase-8- and Bid-dependent pathways. Eur. J. Pharmacol. 2014, 730, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, R.M., Jr. Myocardial protection in heart surgery. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Tibbs, D.; McDonald, L.K.; Lowe, R.F.; Hewitt, R.L. 10% Soybean oil emulsion as a myocardial energy substrate after ischemic arrest. Surg. Forum 1977, 28, 284–285. [Google Scholar] [PubMed]
- Cave, G.; Harvey, M. Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: A systematic review. Acad. Emerg. Med. 2009, 16, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, G.L.; VadeBoncouer, T.; Ramaraju, G.A.; Garcia-Amaro, M.F.; Cwik, M.J. Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats. Anesthesiology 1998, 88, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P.; Morley, P.T.; Vanden Hoek, T.L.; Hickey, R.W.; Kloeck, W.G.; Billi, J.; Bottiger, B.W.; Okada, K.; Reyes, C.; Shuster, M.; et al. Therapeutic hypothermia after cardiac arrest: An advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation 2003, 108, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Field, J.M.; Hazinski, M.F.; Sayre, M.R.; Chameides, L.; Schexnayder, S.M.; Hemphill, R.; Samson, R.A.; Kattwinkel, J.; Berg, R.A.; Bhanji, F.; et al. Part 1: Executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010, 122, S640–S656. [Google Scholar] [CrossRef] [PubMed]
- Vanden Hoek, T.L.; Shao, Z.; Li, C.; Zak, R.; Schumacker, P.T.; Becker, L.B. Reperfusion injury on cardiac myocytes after simulated ischemia. Am. J. Physiol. 1996, 270, H1334–H1341. [Google Scholar] [CrossRef] [PubMed]
- Willert, C.E.; Gharib, M. Digital particle image velocimetry. Exp. Fluids 1991, 10, 181–193. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Z.; Chen, S.-J.; Zhu, X.; Lee, C.; Huang, H.-H.; Meliton, A.; Li, C.; Hoek, T.L.V.; Li, J. Baicalein Rescues Delayed Cooling via Preservation of Akt Activation and Akt-Mediated Phospholamban Phosphorylation. Int. J. Mol. Sci. 2018, 19, 973. https://doi.org/10.3390/ijms19040973
Shao Z, Chen S-J, Zhu X, Lee C, Huang H-H, Meliton A, Li C, Hoek TLV, Li J. Baicalein Rescues Delayed Cooling via Preservation of Akt Activation and Akt-Mediated Phospholamban Phosphorylation. International Journal of Molecular Sciences. 2018; 19(4):973. https://doi.org/10.3390/ijms19040973
Chicago/Turabian StyleShao, Zuohui, Sy-Jou Chen, Xiangdong Zhu, Chunpei Lee, Hsien-Hao Huang, Angelo Meliton, Changqing Li, Terry L. Vanden Hoek, and Jing Li. 2018. "Baicalein Rescues Delayed Cooling via Preservation of Akt Activation and Akt-Mediated Phospholamban Phosphorylation" International Journal of Molecular Sciences 19, no. 4: 973. https://doi.org/10.3390/ijms19040973