Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications

 ,
,  and
and
Abstract
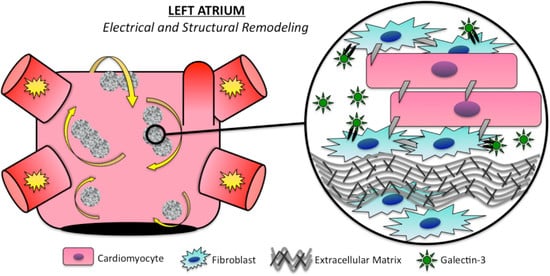
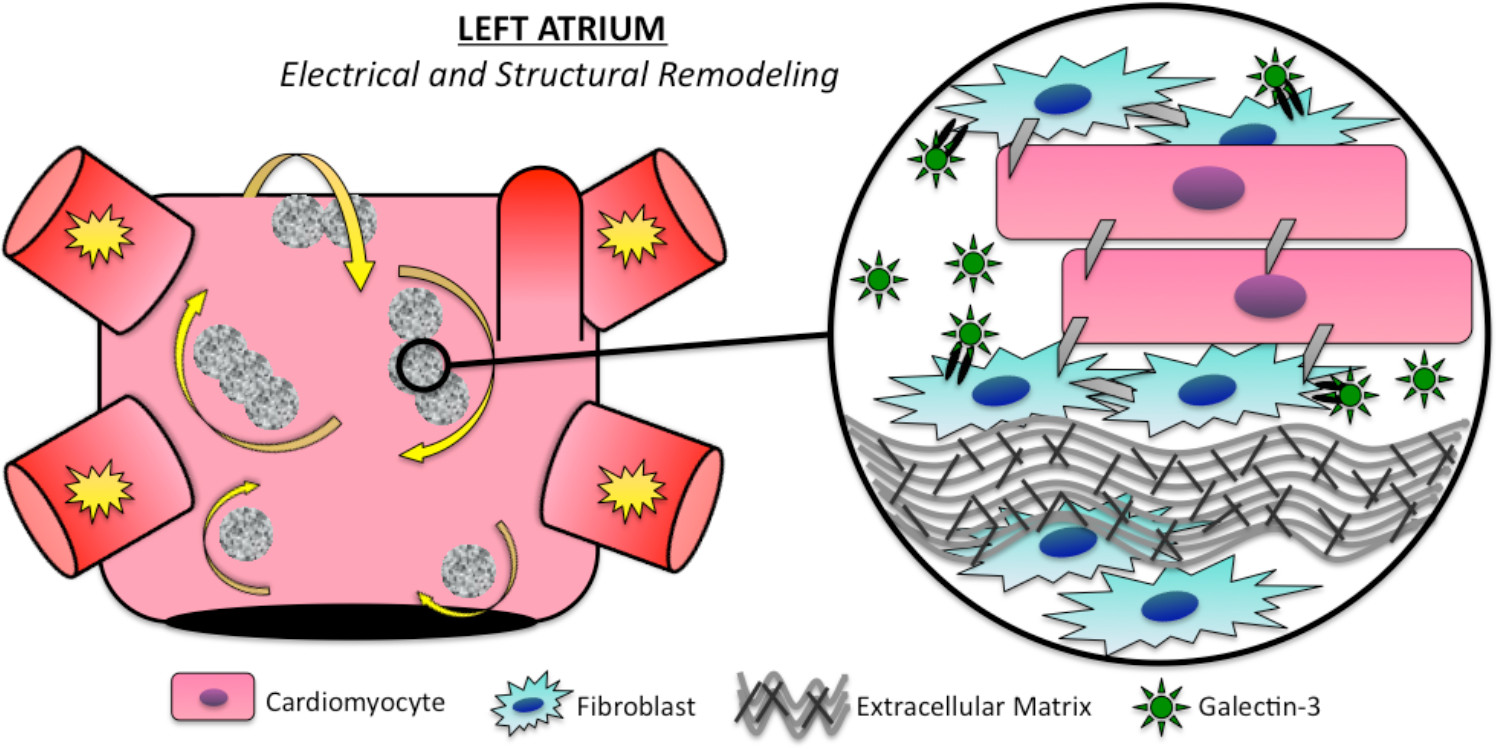
:
{kind=link}
{kind=link}
{kind=link}
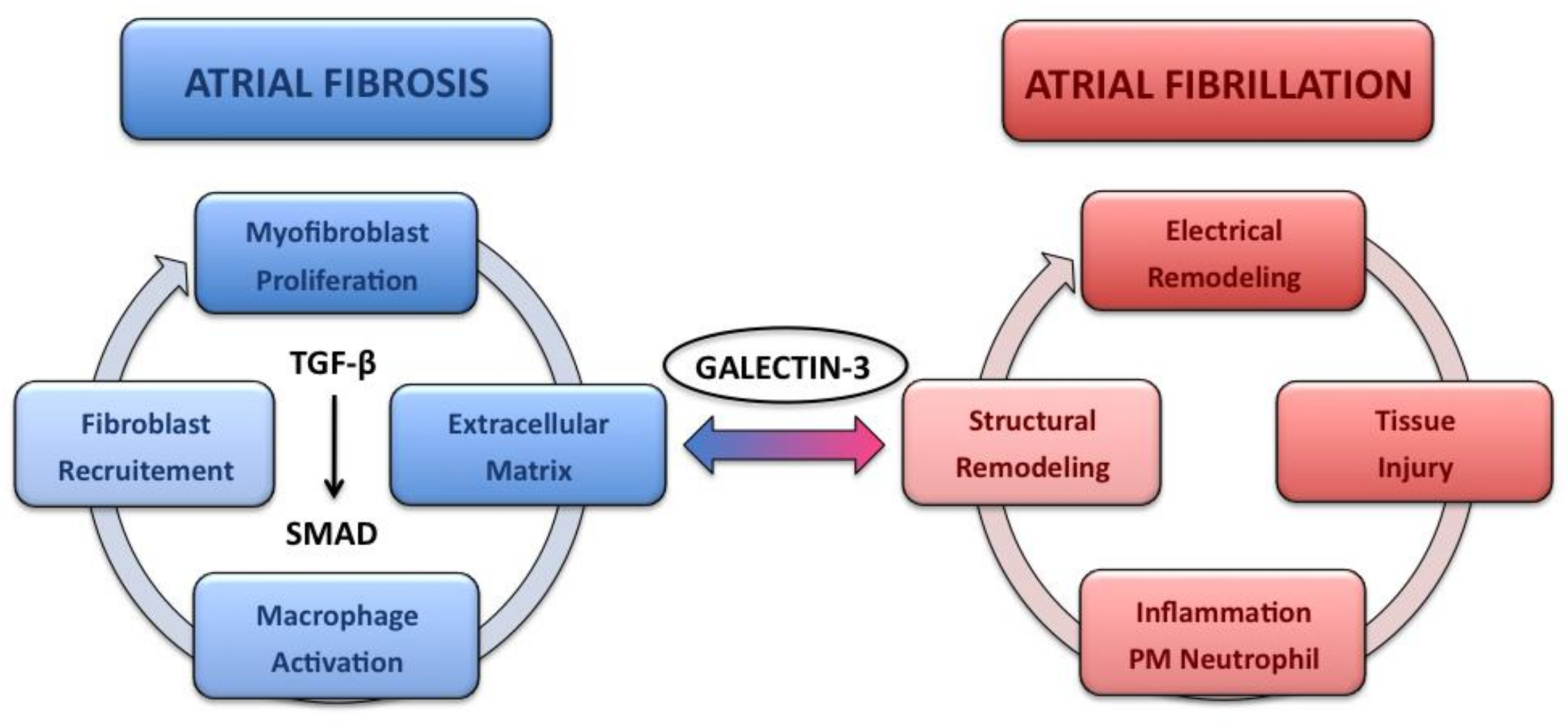{kind=link}
1. Introduction
2. Clinical Atrial Fibrillation
3. Atrial Fibrillation Mechanisms
3.1. Electrical Remodeling
3.2. Structural Remodeling
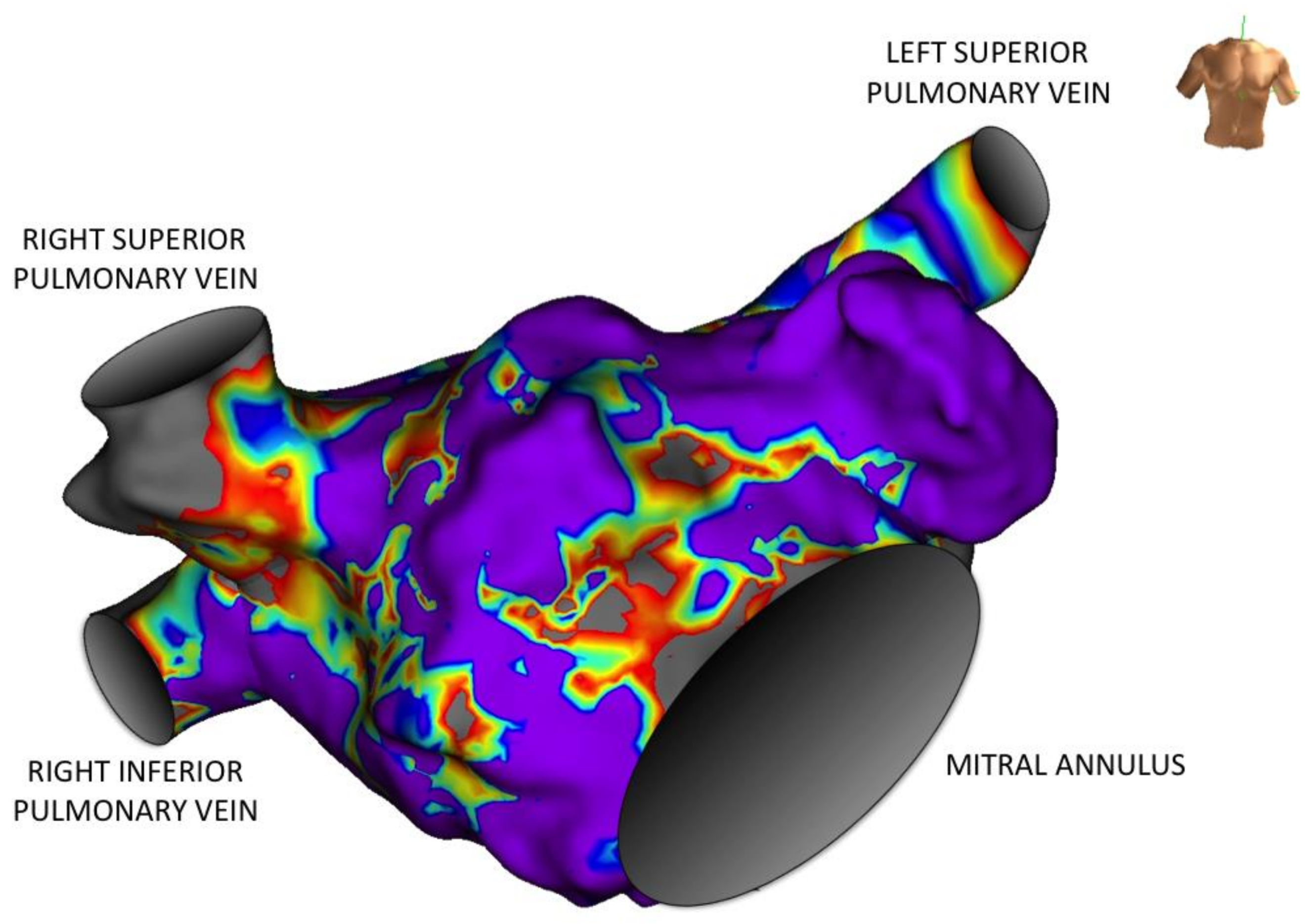
4. Atrial Remodeling and Biomarkers
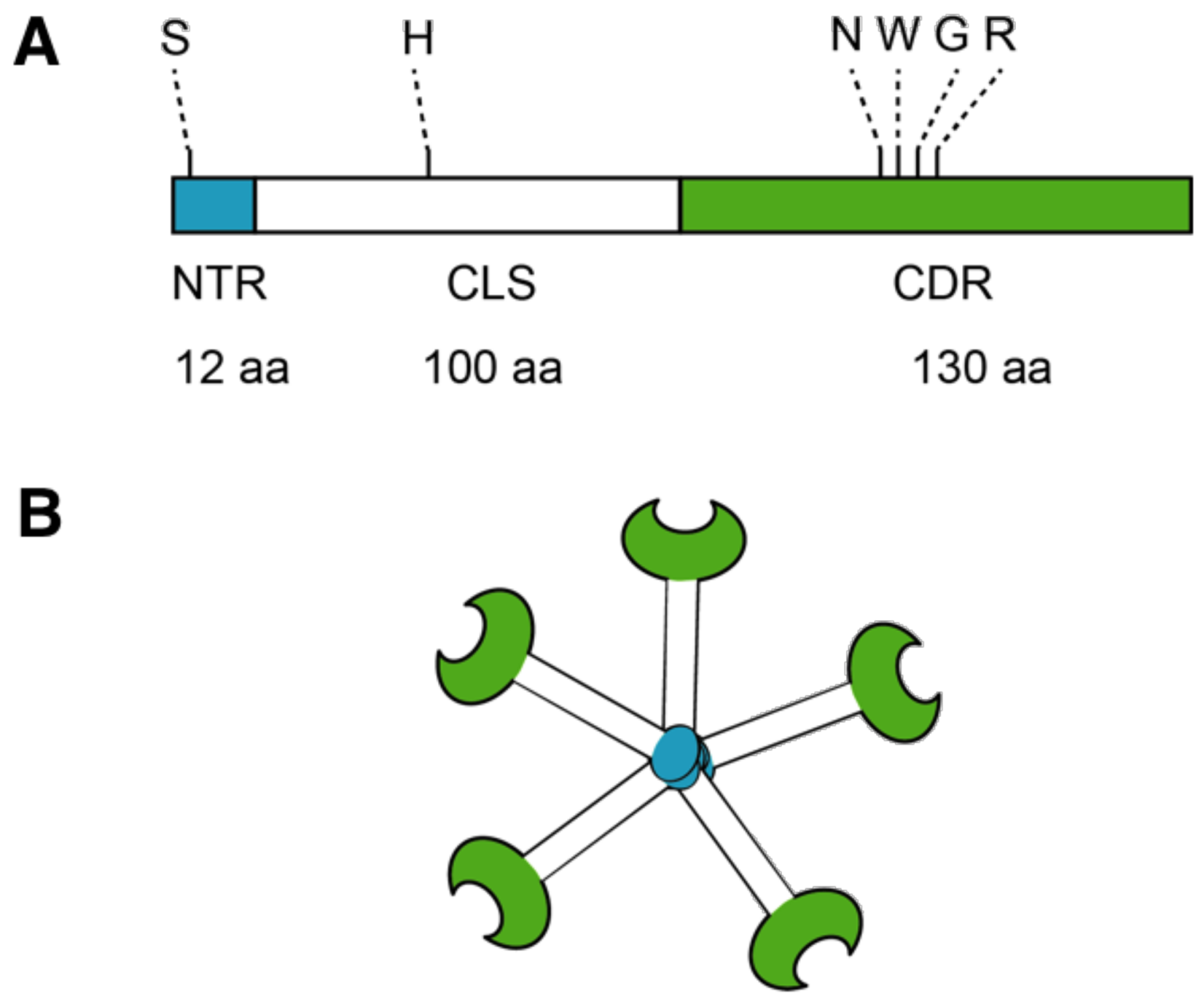
5. Galectin-3 Profibrotic Signaling
6. Disease Progression and Therapeutic Implications
7. Galectin-3 and Atrial Fibrillation Ablation
8. Limitations
9. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| Gal-3 | Galectin-3 |
| MRI | Magnetic resonance imaging |
References
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Goldberger, J.J.; Arora, R.; Green, D.; Greenland, P.; Lee, D.; Lloyd-Jones, D.M.; Marl, M.; Ng, J.; Shah, S.J. Evaluating the atrial myopathy underlying atrial fibrillation: Identifying the arrhythmogenic and thrombogenic substrate. Circulation 2015, 132, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Melgaard, L.; Rasmussen, L.H.; Skjøth, F.; Lip, G.Y.; Larsen, T.B. Age Dependence of RiskFactors for Stroke and Death in Young Patients With Atrial Fibrillation: A NationwideStudy. Stroke 2014, 45, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart J. 2016, 37, 2893–2962. [Google Scholar] [CrossRef] [PubMed]
- Jalife, J. Mechanisms of persistent atrial fibrillation. Curr. Opin. Cardiol. 2014, 29, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Harada, M. Atrial Remodeling and Atrial Fibrillation: Recent Advances and Translational Perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation. A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef] [PubMed]
- Haïssaguerre, M.; Jaïs, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Métayer, P.; Clémenty, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998, 339, 659–566. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.L.; Fisbein, M.C.; Chen, L.S.; Chen, P.S.; Masroor, S. Histopathological substrate for chronic atrial fibrillation in humans. Heart Rhythm. 2009, 6, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quintana, D.; López-Mínguez, J.R.; Pizarro, G.; Murillo, M.; Cabrera, J.A. Triggers and anatomical substrates in the genesis and perpetuation of atrial fibrillation. Curr. Cardiol. Rev. 2012, 8, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Stiles, M.K.; John, B.; Wong, C.X.; Kuklik, P.; Brooks, A.G.; Lau, D.H.; Dimitri, H.; Roberts-Thomson, K.C.; Wilson, L.; De Sciscio, P.; et al. Paroxysmal lone atrial fibrillation is associated with an abnormal atrial substrate: Characterizing the “second factor”. J. Am. Coll. Cardiol. 2009, 53, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.W.; Kistler, P.M.; Lee, G.; Medi, C.; Heck, P.M.; Spence, S.J.; Sparks, P.B.; Morton, J.B.; Kalman, J.M. Electroanatomic remodeling of the left atrium in paroxysmal and persistent atrial fibrillation patients without structural heart disease. J. Cardiovasc. Electrophysiol. 2012, 23, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 2004, 109, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.P.; Kaur, K.; Hwang, E.; Ramirez, R.J.; Willis, B.C.; Filgueiras-Rama, D.; Ennis, S.R.; Takemoto, Y.; Ponce-Balbuena, D.; Zarzoso, M.; et al. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 2014, 129, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Wit, A.L. Is there a role for remodeled connexins in AF? No simple answers. J. Mol. Cell Cardiol. 2008, 44, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, J.V.; Sih, H.J.; Winkle, W.; Zipes, D.P.; Hutchins, G.D.; Olgin, J.E. Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation 2000, 101, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Schotten, U.; Neuberger, H.R.; Bohm, M.; Wirth, K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm. 2011, 8, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Van Hunnik, A.; Hohl, M.; Mahfoud, F.; Wolf, M.; Neuberger, H.R.; Casadei, B.; Reilly, S.N.; Verheule, S.; Bohm, M.; et al. Catheter-based renal denervation reduces atrial nerve sprouting and complexity of atrial fibrillation in goats. Circ. Arrhythm. Electrophysiol. 2015, 8, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Marrouche, N.F.; Wilber, D.; Hindricks, G.; Jais, P.; Akoum, N.; Marchlinski, F.; Kholmovski, E.; Burgon, N.; Hu, N.; Mont, L.; et al. Association of atrial tissue fibrosisidentified by delayedenhancement MRI and atrial fibrillation catheter ablation: The DECAAF study. JAMA 2014, 311, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Jadidi, A.S.; Cochet, H.; Shah, A.J.; Kim, S.J.; Duncan, E.; Miyazaki, S.; Sermesant, M.; Lehrmann, H.; Lederlin, M.; Linton, N.; et al. Inverse relationship between fractionated electrograms and atrial fibrosis in persistentatrial fibrillation: Combined magnetic resonance imaging and high density mapping. J. Am. Coll. Cardiol. 2013, 62, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Malcolme-Lawes, L.C.; Juli, C.; Karim, R.; Bai, W.; Quest, R.; Lim, P.B.; Jamil-Copley, S.; Kojodjojo, P.; Ariff, B.; Davies, D.W.; et al. Automated analysis of atrial late gadolinium enhancement imaging that correlates with endocardial voltage and clinical outcomes: A 2-center study. Heart Rhythm. 2013, 10, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapa, S.; Desjardins, B.; Callans, D.J.; Marchlinski, F.E.; Dixit, S. Contact electroanatomic mapping derived voltage criteria for characterizing left atrial scar in patients undergoing ablation for atrial fibrillation. J. Cardiovasc. Electrophysiol. 2014, 25, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Kottkamp, H. Human atrial fibrillation substrate: Towards a specific fibrotic atrial cardiomyopathy. Eur. Heart J. 2013, 34, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Rolf, S.; Kircher, S.; Arya, A.; Eitel, C.; Sommer, P.; Richter, S.; Gaspar, T.; Bollmann, A.; Altmann, D.; Piedra, C.; Hindricks, G.; Piorkowski, C. Tailored atrial substrate modification based on low-voltage areas in catheter ablation of atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2014, 7, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Arentz, T.; Lehrmann, H.; Sorrel, J.; Markstein, V.; Park, C.; Allgeier, J.; Weber, R.; Jadidi, A. Selective substrate based ablation compared to pulmonary vein isolation alone in patients with persistent atrial fibrillation. Heart Rhythm. 2015, 12, S347. [Google Scholar]
- Haissaguerre, M.; Shah, A.J.; Cochet, H.; Hocini, M.; Dubois, R.; Efimov, I.; Vigmond, E.; Bernus, O.; Trayanova, N. Intermittent drivers anchoring to structural heterogeneities as a major pathophysiological mechanism of human persistent atrial fibrillation. J. Physiol. 2016, 594, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Comtois, P.; Nattel, S. Interactions between cardiac fibrosis spatial pattern and ionic remodeling on electrical wave propagation. In Proceedings of the 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Boston, MA, USA, 30 August–3 September 2011; pp. 4669–4672. [Google Scholar]
- Alonso, S.; Bär, M. Reentry near the percolation threshold in a heterogeneous discrete model for cardiac tissue. Phys. Rev. Lett. 2013, 110, 158101. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Takayama, N.; Shimamoto, T. Directional difference of conduction velocity in the cardiac ventricular syncytium studied by microelectrodes. Circ. Res. 1959, 7, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Valderrábano, M. Influence of anisotropic conduction properties in the propagation of the cardiac action potential. Prog. Biophys. Mol. Biol. 2007, 94, 144–168. [Google Scholar] [CrossRef] [PubMed]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 2015, 36, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Pellman, J.; Lyon, R.C.; Sheikh, F. Extracellular matrix remodeling in atrial fibrosis: Mechanisms and implications in atrial fibrillation. J. Mol. Cell. Cardiol. 2010, 48, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, I.; Szapáry, L.; Csécsei, P.; Csanádi, Z.; Csiba, L. Potential Biological Markers of Atrial Fibrillation: A Chance to Prevent Cryptogenic Stroke. BioMed Res. Int. 2017, 2017, 8153024. [Google Scholar] [CrossRef] [PubMed]
- Van Den Steen, P.E.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 2002, 37, 375–536. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, G.; Xie, B.; Babu, K.; Huang, C. Changes in matrix metalloproteinase-9 levels during progression of atrial fibrillation. J. Int. Med. Res. 2014, 42, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Liu, H.; Ng, C.Y.; Xu, G.; Liu, E.; Li, G.; Liu, T. Circulating serum levels of growth differentiation factor-15 and neuregulin-1 in patients with paroxysmal non-valvular atrial fibrillation. Int. J. Cardiol. 2014, 172, e311–e313. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.A.; Maziarz, M.; Tan, A.Y.; Glazer, N.L.; Zieman, S.J.; Kizer, J.R.; Ix, J.H.; Djousse, L.; Siscovick, D.S.; Heckbert, S.R.; et al. Circulating fibrosis biomarkers and risk of atrial fibrillation: The Cardiovascular Health Study (CHS). Am. Heart J. 2014, 167, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Funasaka, T.; Raz, A.; Nangia-Makker, P. Nuclear transport of galectin-3 and its therapeutic implications. Semin. Cancer Biol. 2014, 27, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.C. Mac-2: A versatile galactose-binding protein of mammalian tissues. Glycobiology 1994, 4, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.; Cummings, R.D.; Drickamer, K.; Feizi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.; et al. Galectins: A family of animal beta-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Akahani, S.; Nangia-Makker, P.; Inohara, H.; Kim, H.R.; Raz, A. Galectin-3: A novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 1997, 57, 5272–5276. [Google Scholar] [PubMed]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; Ho, J.E.; Liu, F.T.; de Boer, R.A. Galectin-3 activation and inhibition in heart failure and cardiovascular disease: An update. Theranostics 2018, 8, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Li, J.; Gao, J. Functions of galectin-3 and its role in fibrotic diseases. J. Pharmacol. Exp. Ther. 2014, 351, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.C.; Pokharel, S.; van Brakel, T.J.; van Berlo, J.H.; Cleutjens, J.P.; Schroen, B.; André, S.; Crijns, H.J.; Gabius, H.J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell Sci. 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Pauschinger, M.; Knopf, D.; Petschauer, S.; Doerner, A.; Poller, W.; Schwimmbeck, P.L.; Kuhl, U.; Schultheiss, H.P. Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation 1999, 99, 2750–2756. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-beta-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.J.; Li, Y.; Sun, A.J.; Liu, J.J.; Ji, K.D.; Zhang, Y.Z.; Sun, W.L.; Marche, P.; Zhu, D.L. Differentiation of vascular myofibroblasts induced by transforming growth factor-beta1 requires the involvement of protein kinase Calpha. J. Mol. Cell. Cardiol. 2003, 35, 1105–1112. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Chen, W.D.; Zhu, D.L.; Wu, L.Y.; Zhang, J.; Han, W.Q.; Li, J.D.; Yan, C.; Gao, P.J. The PDE1A-PKCalpha signaling pathway is involved in the upregulation of alpha-smooth muscle actin by TGF-beta1 in adventitial fibroblasts. J. Vasc. Res. 2010, 47, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Villar, A.V.; Garcia, R.; Llano, M.; Cobo, M.; Merino, D.; Lantero, A.; Tramullas, M.; Hurle, J.M.; Hurle, M.A.; Nistal, J.F. BAMBI (BMP and activin membrane-bound inhibitor) protects the murine heart from pressure-overload biomechanical stress by restraining TGF-beta signaling. Biochim. Biophys. Acta 2013, 1832, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P.; et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ruifrok, W.P.; Meissner, M.; Bos, E.M.; van Goor, H.; Sanjabi, B.; van der Harst, P.; Pitt, B.; Goldstein, I.J.; Koerts, J.A.; et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ. Heart Fail. 2013, 6, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 2013, 8, e83481. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Gabius, H.J.; André, S.; Kaltner, H.; Sabesan, S.; Roy, R.; Liu, B.; Macaluso, F.; Brewer, C.F. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J. Biol. Chem. 2004, 279, 10841–10847. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y.; Ramirez, R.J.; Yokokawa, M.; Kaur, K.; Ponce-Balbuena, D.; Sinno, M.C.; Willis, B.C.; Ghanbari, H.; Ennis, S.R.; Guerrero-Serna, G.; et al. Galectin-3 Regulates Atrial Fibrillation Remodeling and Predicts Catheter Ablation Outcomes. JACC Basic. Transl. Sci. 2016, 1, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.V.; Fagard, R.H.; Lijnen, P.J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 2002, 39, 258–263. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; van Veldhuisen, D.J.; Gansevoort, R.T.; Muller Kobold, A.C.; van Gilst, W.H.; Hillege, H.L.; Bakker, S.J.; van der Harst, P. The fibrosis marker galectin-3 and outcome in the general population. J. Intern. Med. 2012, 272, 55–64. [Google Scholar] [CrossRef] [PubMed]
- De Couto, G.; Ouzounian, M.; Liu, P.P. Early detection of myocardial dysfunction and heart failure. Nat. Rev. Cardiol. 2010, 7, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.L., Jr.; Kiernan, M.S.; et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e1054–e1091. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.E.; Yin, X.; Levy, D.; Vasan, R.S.; Magnani, J.W.; Ellinor, P.T.; McManus, D.D.; Lubitz, S.A.; Larson, M.G.; Benjamin, E.J. Galectin 3 and incident atrial fibrillation in the community. Am. Heart J. 2014, 167, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Clementy, N.; Piver, E.; Benhenda, N.; Bernard, A.; Pierre, B.; Simeon, E.; Fauchier, L.; Pages, J.C.; Babuty, D. Galectin-3 in patients undergoing ablation of atrial fibrillation. IJC Metabolic. Endocrine. 2014, 5, 56–60. [Google Scholar] [CrossRef]
- Szadkowska, I.; Wlazeł, R.N.; Migała, M.; Szadkowski, K.; Zielińska, M.; Paradowski, M.; Pawlicki, L. The association between galectin-3 and clinical parameters in patients with first acute myocardial infarction treated with primary percutaneous coronary angioplasty. Cardiol. J. 2013, 20, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, M.U.; Gurses, K.M.; Kocyigit, D.; Canpinar, H.; Canpolat, U.; Evranos, B.; Yorgun, H.; Sahiner, M.L.; Kaya, E.B.; Hazirolan, T.; et al. The Association of Serum Galectin-3 Levels with Atrial Electrical and Structural Remodeling. J. Cardiovasc. Electrophysiol. 2015, 26, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Wachtell, K.; Lehto, M.; Gerdts, E.; Olsen, M.H.; Hornestam, B.; Dahlöf, B.; Ibsen, H.; Julius, S.; Kjeldsen, S.E.; Lindholm, L.H.; et al. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: The Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J. Am. Coll. Cardiol. 2005, 45, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Edsfeldt, A.; Bengtsson, E.; Asciutto, G.; Dunér, P.; Björkbacka, H.; Fredrikson, G.N.; Nilsson, J.; Goncalves, I. High plasma levels of Galectin-3 are associated with increased risk for stroke after carotid endarterectomy. Cerebrovasc. Dis. 2016, 41, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Z.H.; Zhang, N.; Liu, S.D.; Zhao, J.J.; Liu, S.Y. Serum Galectin-3 level, not Galectin-1, is associated with the clinical feature and outcome in patients with acute ischemic stroke. Oncotarget 2017, 8, 109752–109761. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhong, C.; Zhu, Z.; Xu, T.; Peng, Y.; Xu, T.; Peng, H.; Chen, C.S.; Wang, J.; Ju, Z.; et al. Serum Galectin-3 and Poor Outcomes Among Patients With Acute Ischemic Stroke. Stroke 2018, 49, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Asberg, S.; Hijazi, Z.; Siegbahn, A.; Andersson, U.; Granger, C.B.; Hanna, M.; Horowitz, J.D.; Lopes, R.D.; Lindahl, B.; Wallentin, L. Galectin-3 is associated with worse clinical outcome in patients with atrial fibrillation: A substudy from the ARISTOTLE trial. Eur. Heart J. 2014, 35, 426. [Google Scholar]
- Verma, A.; Mantovan, R.; Macle, L.; De Martino, G.; Chen, J.; Morillo, C.A.; Novak, P.; Calzolari, V.; Guerra, P.G.; Nair, G.; et al. Substrate and Trigger Ablation for Reduction of Atrial Fibrillation (STAR AF): A randomized, multicentre, international trial. Eur. Heart J. 2010, 31, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Hindricks, G.; Shoemaker, M.B.; Husser, D.; Arya, A.; Sommer, P.; Rolf, S.; Saavedra, P.; Kanagasundram, A.; Patrick Whalen, S.; et al. The APPLE score: A novel and simple score for the prediction of rhythm outcomes after catheter ablation of atrial fibrillation. Clin. Res. Cardiol. 2015, 104, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Li, S.N.; Wen, S.N.; Nie, J.G.; Deng, W.N.; Bai, R.; Liu, N.; Tang, R.B.; Zhang, T.; Du, X.; et al. Plasma galectin-3 predicts clinical outcomes after catheter ablation in persistent atrial fibrillation patients without structural heart disease. Europace 2015, 17, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Clementy, N.; Benhenda, N.; Piver, E.; Pierre, B.; Bernard, A.; Fauchier, L.; Pages, J.C.; Babuty, D. Serum Galectin-3 Levels Predict Recurrences after Ablation of Atrial Fibrillation. Sci. Rep. 2016, 6, 34357. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Wang, Y.; Tang, K.; Li, X.; Peng, W.; Liang, C.; Xu, Y. Association between left atrial size and atrial fibrillation recurrence after single circumferential pulmonary vein isolation: A systematic review and meta-analysis of observational studies. Europace 2012, 14, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Sramko, M.; Peichl, P.; Wichterle, D.; Tintera, J.; Weichet, J.; Maxian, R.; Pasnisinova, S.; Kockova, R.; Kautzner, J. Clinical value of assessment of left atrial late gadolinium enhancement in patients undergoing ablation of atrial fibrillation. Int. J. Cardiol. 2015, 179, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Schmidl, J.; Bollmann, A. Galectin-3 in atrial fibrillation: A novel marker of atrial remodeling or just bystander? Am. J. Cardiol. 2015, 116, 163. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Mohanty, P.; Di Biase, L.; Bai, R.; Pump, A.; Santangeli, P.; Burkhardt, D.; Gallinghouse, J.G.; Horton, R.; Sanchez, J.E.; et al. Impact of metabolic syndrome on procedural outcomes in patients with atrial fibrillation undergoing catheter ablation. J. Am. Coll. Cardiol. 2012, 59, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Cervellin, G.; Sanchis-Gomar, F. Galectin-3 in atrial fibrillation: Simple bystander, player or both? Clin. Biochem. 2015, 48, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Jiang, C.Y.; Betts, T.R.; Chen, J.; Deisenhofer, I.; Mantovan, R.; Macle, L.; Morillo, C.A.; Haverkamp, W.; Weerasooriya, R.; et al. STAR AF II Investigators. Approaches to catheter ablation for persistent atrial fibrillation. N. Engl. J. Med. 2015, 372, 1812–1822. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clementy, N.; Piver, E.; Bisson, A.; Andre, C.; Bernard, A.; Pierre, B.; Fauchier, L.; Babuty, D. Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 976. https://doi.org/10.3390/ijms19040976
Clementy N, Piver E, Bisson A, Andre C, Bernard A, Pierre B, Fauchier L, Babuty D. Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications. International Journal of Molecular Sciences. 2018; 19(4):976. https://doi.org/10.3390/ijms19040976
Chicago/Turabian StyleClementy, Nicolas, Eric Piver, Arnaud Bisson, Clémentine Andre, Anne Bernard, Bertrand Pierre, Laurent Fauchier, and Dominique Babuty. 2018. "Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications" International Journal of Molecular Sciences 19, no. 4: 976. https://doi.org/10.3390/ijms19040976
APA StyleClementy, N., Piver, E., Bisson, A., Andre, C., Bernard, A., Pierre, B., Fauchier, L., & Babuty, D. (2018). Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications. International Journal of Molecular Sciences, 19(4), 976. https://doi.org/10.3390/ijms19040976