Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats


 ,
,
Abstract
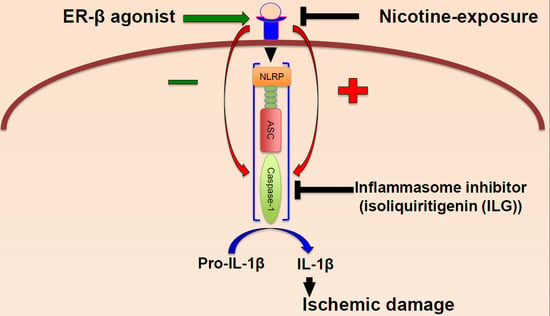
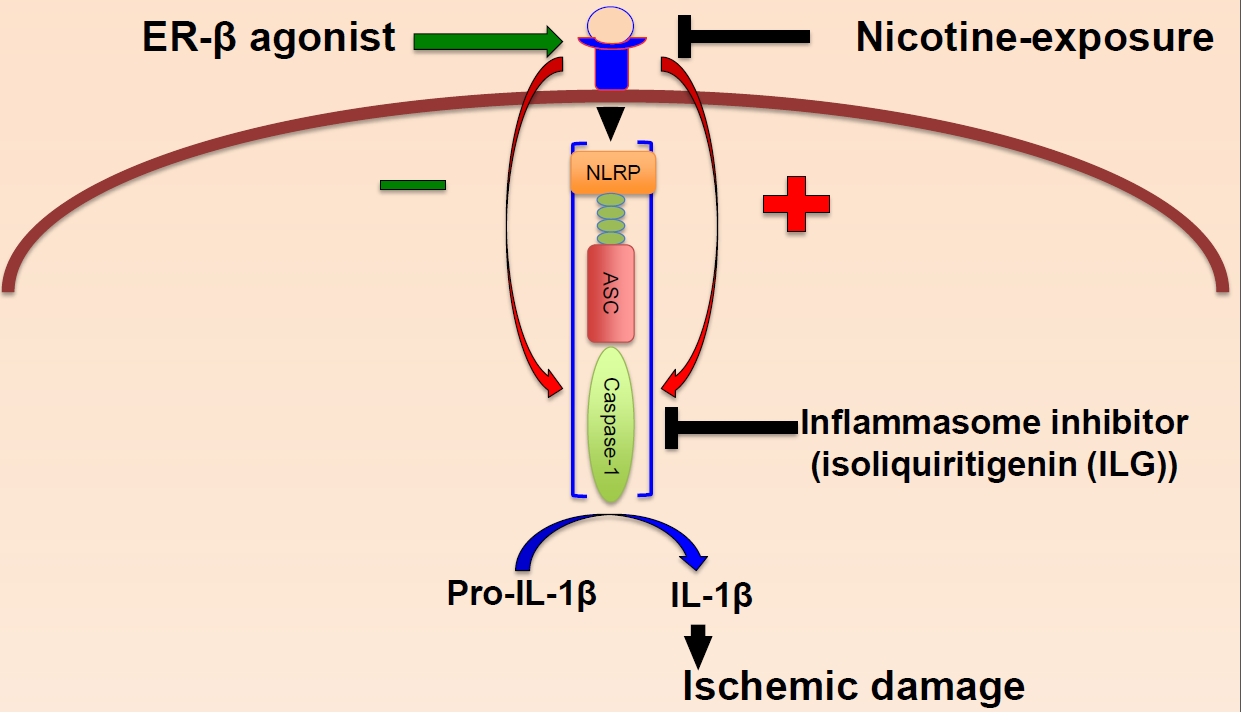
:
1. Introduction
2. Results
2.1. Nicotine Reduces ER-β Protein Levels in the Brain of Female Rats
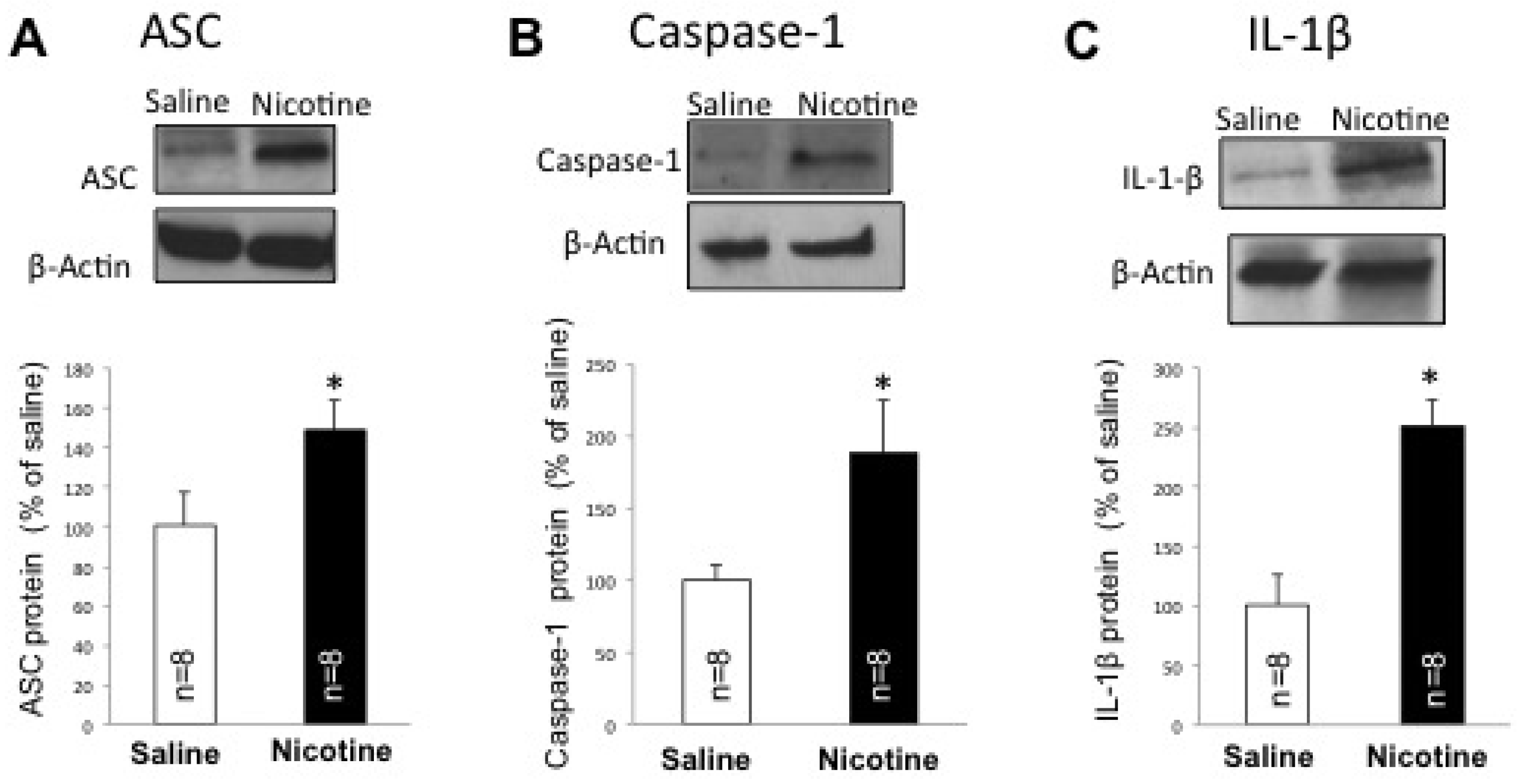
2.2. Nicotine Increases Inflammasome Activation in the Brain of Female Rats
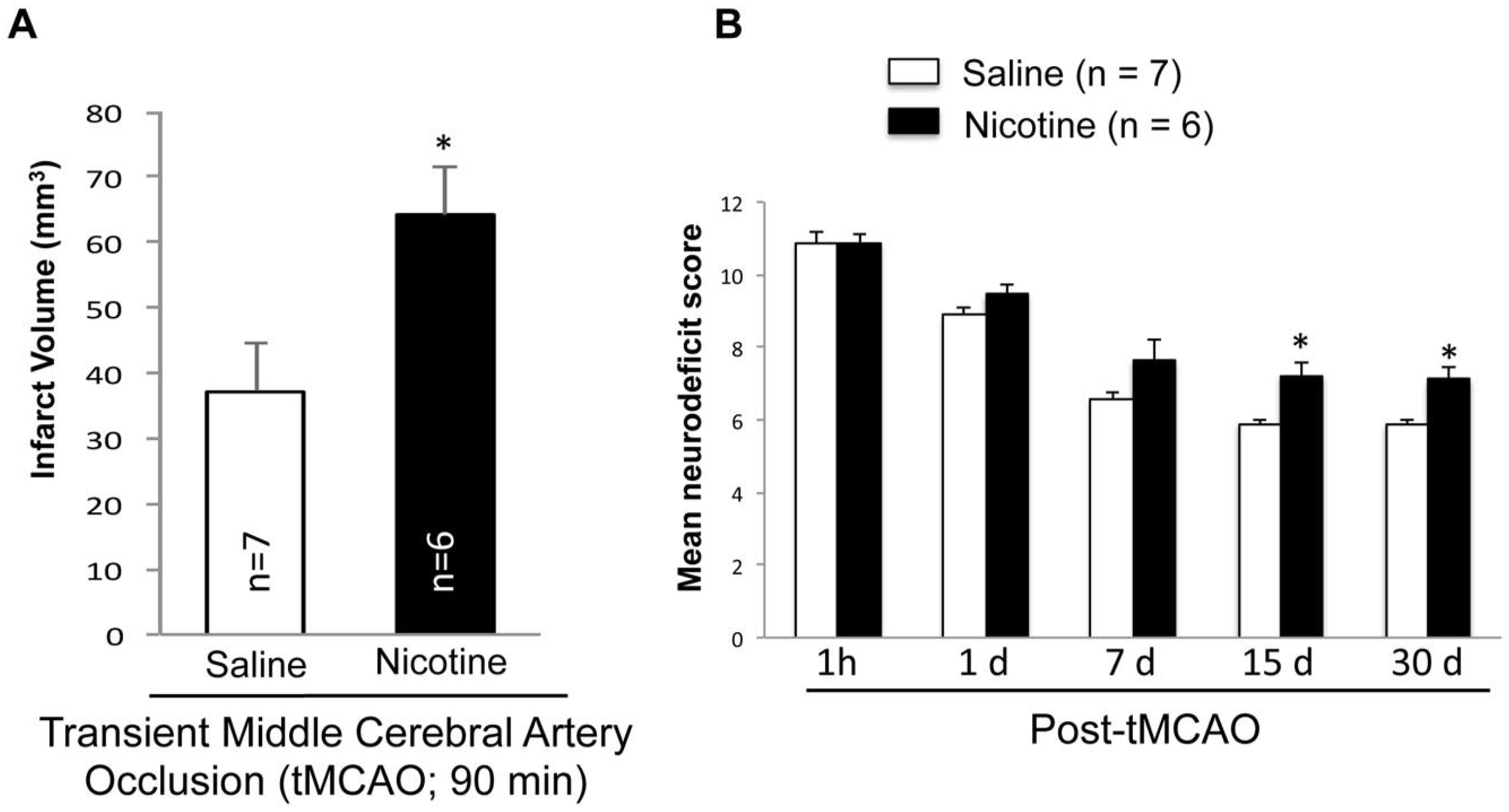
2.3. Nicotine Worsens Infarct Volume and Neurodeficit Score after tMCAO
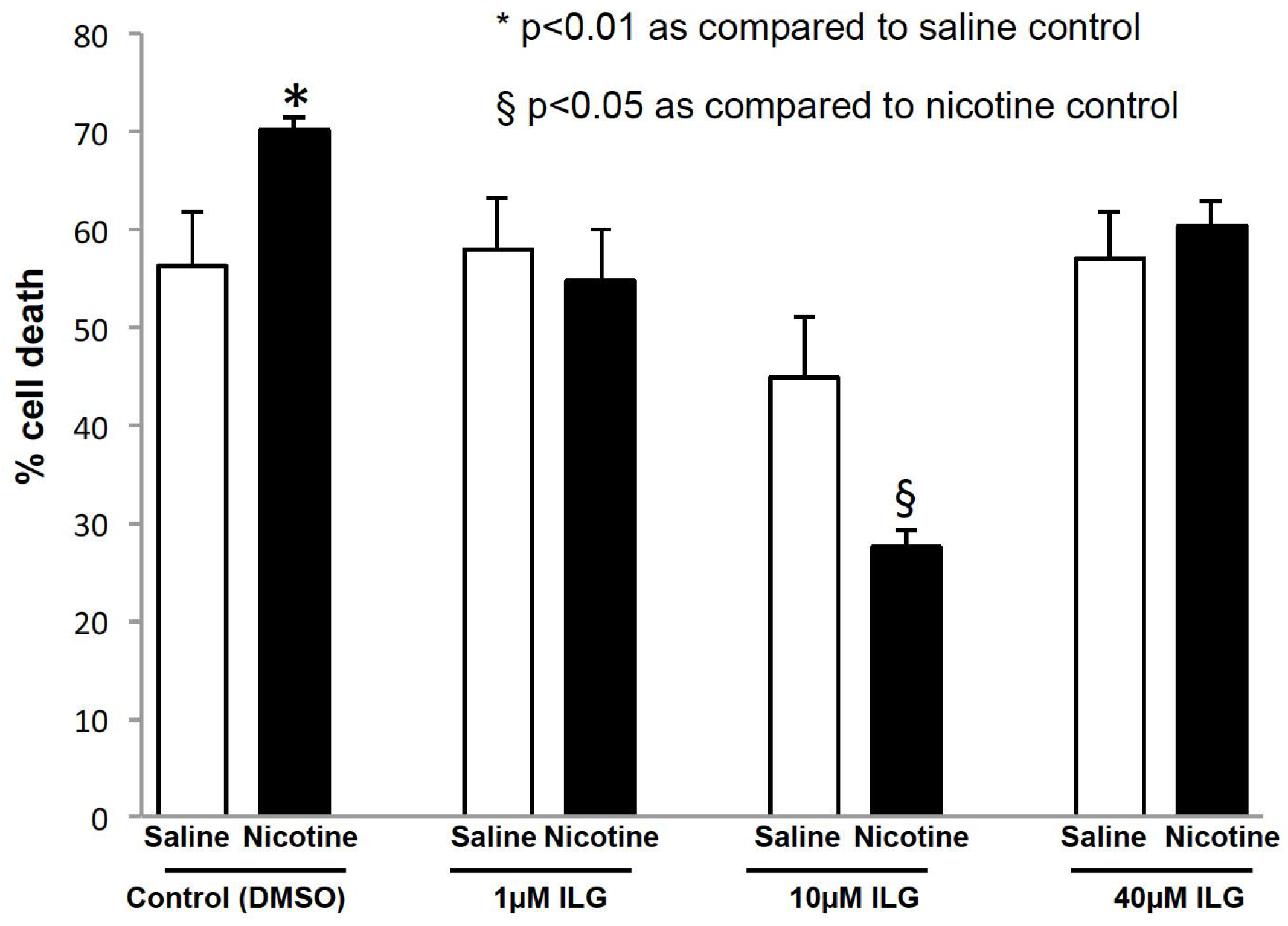
2.4. Inhibition of Inflammasome Activation Decreases Neuronal Cell Death in an In Vitro Model of Stroke
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. In Vivo
4.3. Nicotine or Saline Treatment
4.4. Transient Middle Cerebral Artery Occlusion (tMCAO) and Infarct Volume
4.5. Neurodeficit Scoring
4.6. Western Blotting
4.7. In Vitro Organotypic Slice Cultures and Oxygen-Glucose Deprivation
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ASC | Apoptosis associated speck-like protein containing a caspase recruitment domain |
| BBB | Blood brain barrier |
| DMSO | Dimethyl sulfoxide |
| e-Cigarette | Electronic cigarettes |
| ER-β | Estrogen receptor subtype beta |
| FAM | Formaldehyde, glacial acetic acid, and methanol |
| IL-1β | Interleukin-1β |
| ILG | Isoliquiritigenin |
| nAChR | Nicotinic acetylcholine receptor |
| NLR | NOD-like receptor |
| NMDA | N-methyl-d-aspartate |
| OGD | Oxygen-glucose deprivation |
| PI | Propidium iodide |
| tMCAO | Transient middle cerebral artery occlusion |
| TNF | Tumor necrosis factor |
References
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Huxley, R.R.; Woodward, M. Smoking as a risk factor for stroke in women compared with men: A systematic review and meta-analysis of 81 cohorts, including 3,980,359 individuals and 42,401 strokes. Stroke 2013, 44, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Loraine, A.; West, S.C.; Goodkind, D.; He, W. 65+ in the United States: 2010. Curr. Popul. Rep. 2014, 23–212. [Google Scholar]
- Girijala, R.L.; Sohrabji, F.; Bush, R.L. Sex differences in stroke: Review of current knowledge and evidence. Vasc. Med. 2017, 22, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Morris, J.; Pikula, A. Stroke prevention: Modifying risk factors. Ther. Adv. Cardiovasc. Dis. 2008, 2, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Uchino, K. Treatment of risk factors to prevent stroke. Neurotherapeutics 2011, 8, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, A.; Lamassa, M.; Baldereschi, M.; Pracucci, G.; Basile, A.M.; Wolfe, C.D.; Giroud, M.; Rudd, A.; Ghetti, A.; Inzitari, D. Sex differences in the clinical presentation, resource use, and 3-month outcome of acute stroke in Europe: Data from a multicenter multinational hospital-based registry. Stroke 2003, 34, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Hirsch, N.; Dave, K.R.; Yavagal, D.R.; Bramlett, H.; Saul, I. Nicotine and estrogen synergistically exacerbate cerebral ischemic injury. Neuroscience 2011, 181, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Rune, G.M.; Frotscher, M. Neurosteroid synthesis in the hippocampus: Role in synaptic plasticity. Neuroscience 2005, 136, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, R.L.; Gochberg, J.; Ryan, K.J. Nicotine, cotinine, and anabasine inhibit aromatase in human trophoblast in vitro. J. Clin. Investig. 1986, 77, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Cassidenti, D.L.; Vijod, A.G.; Vijod, M.A.; Stanczyk, F.Z.; Lobo, R.A. Short-term effects of smoking on the pharmacokinetic profiles of micronized estradiol in postmenopausal women. Am. J. Obstet. Gynecol. 1990, 163, 1953–1960. [Google Scholar] [CrossRef]
- Cramer, D.W.; Harlow, B.L.; Xu, H.; Fraer, C.; Barbieri, R. Cross-sectional and case-controlled analyses of the association between smoking and early menopause. Maturitas 1995, 22, 79–87. [Google Scholar] [CrossRef]
- Grainge, M.J.; Coupland, C.A.C.; Cliffe, S.J.; Chilvers, C.E.D.; Hosking, D.J.; Nottingham EPIC Study Group. Cigarette smoking, alcohol and caffeine consumption, and bone mineral density in postmenopausal women. Osteoporos. Int. 1998, 8, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, G.; Thompson, S.G.; Meade, T.W. Relation between cigarette smoking and use of hormonal replacement therapy for menopausal symptoms. J. Epidemiol. Community Health 1987, 41, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Christiansen, C.; Rodbro, P. Cigarette smoking, serum estrogens, and bone loss during hormone-replacement therapy early after menopause. N. Engl. J. Med. 1985, 313, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Michnovicz, J.J.; Naganuma, H.; Hershcopf, R.J.; Bradlow, H.L.; Fishman, J. Increased urinary catechol estrogen excretion in female smokers. Steroids 1988, 52, 69–83. [Google Scholar] [CrossRef]
- Mueck, A.O.; Seeger, H. Smoking, estradiol metabolism and hormone replacement therapy. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2005, 3, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Windham, G.C.; Elkin, E.P.; Swan, S.H.; Waller, K.O.; Fenster, L. Cigarette smoking and effects on menstrual function. Obstet. Gynecol. 1999, 93, 59–65. [Google Scholar] [PubMed]
- Zhang, Q.G.; Raz, L.; Wang, R.; Han, D.; De Sevilla, L.; Yang, F.; Vadlamudi, R.K.; Brann, D.W. Estrogen attenuates ischemic oxidative damage via an estrogen receptor α-mediated inhibition of NADPH oxidase activation. J. Neurosci. 2009, 29, 13823–13836. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B.; Rau, S.W.; Shughrue, P.J.; Zhu, H.; Yu, J.; Cashion, A.B.; Suzuki, S.; Gerhold, L.M.; Bottner, M.B.; Dubal, S.B. Differential modulation of estrogen receptors (ERs) in ischemic brain injury: A role for ER-α in estradiol-mediated protection against delayed cell death. Endocrinology 2006, 147, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Lebesgue, D.; Chevaleyre, V.; Zukin, R.S.; Etgen, A.M. Estradiol rescues neurons from global ischemia-induced cell death: Multiple cellular pathways of neuroprotection. Steroids 2009, 74, 555–561. [Google Scholar] [CrossRef] [PubMed]
- De Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Patel, H.H.; Brand, F.J.; Perez-Pinzon, M.A.; Bramlett, H.M.; Raval, A.P. Estrogen receptor β signaling alters cellular inflammasomes activity after global cerebral ischemia in reproductively senescence female rats. J. Neurochem. 2016, 136, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Dave, K.R.; Saul, I.; Gonzalez, G.J.; Diaz, F. Synergistic inhibitory effect of nicotine plus oral contraceptive on mitochondrial complex-IV is mediated by estrogen receptor β in female rats. J. Neurochem. 2012, 121, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflammation 2017, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.X.; de Oliveira, G.V.; de Araújo, F.Y.R.; de Barros Viana, G.S.; de Sousa, F.C.F.; Hyphantis, T.N.; Grunberg, N.E.; Carvalho, A.F.; Macêdo, D.S. Differences in vulnerability to nicotine-induced kindling between female and male periadolescent rats. Psychopharmacology 2013, 225, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Zhang, X.; Wang, T.; Liang, L.; Chang, W.; Xia, H. Pharmacokinetics, biodistribution and bioavailability of isoliquiritigenin after intravenous and oral administration. Pharm. Biol. 2014, 52, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Sick, J.T.; Gonzalez, G.J.; DeFazio, R.A.; Dong, C.; Sick, T.J. Chronic nicotine exposure inhibits estrogen-mediated synaptic functions in hippocampus of female rats. Neurosci. Lett. 2012, 517, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Yildirim, M.; Janssen, W.G.; Lou, W.W.; McEwen, B.S.; Morrison, J.H.; Milner, T.A. Estrogen and aging affect the synaptic distribution of estrogen receptor beta-immunoreactivity in the CA1 region of female rat hippocampus. Brain Res. 2011, 1379, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Hauenstein, A.V.; Zhang, L.; Wu, H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr. Opin. Struct. Biol. 2015, 31, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Kim, J.J.; Kim, T.S.; Lee, P.Y.; Kim, S.Y.; Lee, H.M.; Shin, D.M.; Nguyen, L.T.; Lee, M.S.; Jin, H.S.; et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6115. [Google Scholar] [CrossRef] [PubMed]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Zoli, M. Neuroprotection via nAChRs: The role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Front. Biosci. 2008, 13, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A. Nicotine and the adolescent brain: Insights from an animal model. Neurotoxicol. Teratol. 2002, 24, 369–384. [Google Scholar] [CrossRef]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S.; et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Arch. Eur. J. Physiol. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Barsukova, A.G.; Bourdette, D.; Forte, M. Mitochondrial calcium and its regulation in neurodegeneration induced by oxidative stress. Eur. J. Neurosci. 2011, 34, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Toman, J.; Fiskum, G. Influence of aging on membrane permeability transition in brain mitochondria. J. Bioenerg. Biomembr. 2011, 43, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, F.; Giacomello, M.; Donati, Y.; Carnesecchi, S.; Frieden, M.; Barazzone-Argiroffo, C. Nicotine mediates oxidative stress and apoptosis through cross talk between NOX1 and Bcl-2 in lung epithelial cells. Free Radic. Biol. Med. 2014, 76, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Clark, J.; Reed, D.K.; Juncos, L.A. Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc. Nephrol. Dial. Transplant. 2013, 28, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Vijayasarathy, C.; Raza, H.; Mullick, J.; Avadhani, N.G. Preferential effects of nicotine and 4-(N-methyl-N-nitrosamine)-1-(3-pyridyl)-1-butanone on mitochondrial glutathione S-transferase A4-4 induction and increased oxidative stress in the rat brain. Biochem. Pharmacol. 1998, 56, 831–839. [Google Scholar] [CrossRef]
- Marcondes, F.K.; Bianchi, F.J.; Tanno, A.P. Determination of the estrous cycle phases of rats: Some helpful considerations. Braz. J. Biol. 2002, 62, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Saul, I.; Dave, K.R.; DeFazio, R.A.; Perez-Pinzon, M.A.; Bramlett, H. Pretreatment with a single estradiol-17β bolus activates cyclic-AMP response element binding protein and protects CA1 neurons against global cerebral ischemia. Neuroscience 2009, 160, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.; Raval, A.P.; Dembner, J.M.; Pérez-Pinzón, M.A.; Steinberg, G.K.; Yenari, M.A.; Mochly-Rosen, D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. 2004, 24, 6880–6888. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.M.; Sun, G.H.; Kunis, D.; Yenari, M.A.; Steinberg, G.K. Delayed induction and long-term effects of mild hypothermia in a focal model of transient cerebral ischemia: Neurological outcome and infarct size. J. Neurosurg. 2001, 94, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Ginsberg, M.D. Quantitative assessment of the normal cerebral microvasculature by endothelial barrier antigen (EBA) immunohistochemistry: Application to focal cerebral ischemia. Brain Res. 2000, 865, 237–244. [Google Scholar] [CrossRef]
- Zhang, Y.; Belayev, L.; Zhao, W.; Irving, E.A.; Busto, R.; Ginsberg, M.D. A selective endothelin ETA receptor antagonist, SB 234551, improves cerebral perfusion following permanent focal cerebral ischemia in rats. Brain Res. 2005, 1045, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.; Khoutorova, L.; Zhao, W.; Vigdorchik, A.; Belayev, A.; Busto, R.; Magal, E.; Ginsberg, M.D. Neuroprotective effect of darbepoetin alfa, a novel recombinant erythropoietic protein, in focal cerebral ischemia in rats. Stroke 2005, 36, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Ley, J.J.; Vigdorchik, A.; Belayev, L.; Zhao, W.; Busto, R.; Khoutorova, L.; Becker, D.A.; Ginsberg, M.D. Stilbazulenyl nitrone, a second-generation azulenyl nitrone antioxidant, confers enduring neuroprotection in experimental focal cerebral ischemia in the rat: Neurobehavior, histopathology, and pharmacokinetics. J. Pharmacol. Exp. Ther. 2005, 313, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.P.; Dave, K.R.; Vivero, R.; Schmidt-Kastner, R.; Sick, T.J.; Pérez-Pinzón, M.A. Improvement in neuronal survival after ischemic preconditioning in hippocampal slice cultures. Brain Res. 2002, 952, 153–158. [Google Scholar] [CrossRef]
- Raval, A.P.; Dave, K.R.; Mochly-Rosen, D.; Sick, T.J.; Pérez-Pinzón, M.A. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J. Neurosci. 2003, 23, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Lange-Asschenfeldt, C.; Raval, A.P.; Dave, K.R.; Mochly-Rosen, D.; Sick, T.J.; Pérez-Pinzón, M.A. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J. Cereb. Blood Flow Metab. 2004, 24, 636–645. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Glucose | pH | pCO2 | pO2 | MABP | Post-tMCAO Mortality |
|---|---|---|---|---|---|---|
| Saline + Sham (n = 4) | 131.0 ± 7.4 | 7.39 ± 0.5 | 36.3 ± 2.3 | 135 ± 35 | 132.4 ± 6 | 0 |
| 7.39 ± 0.4 | 36.0 ± 4.1 | 133 ± 15 | 134 ± 11.7 | |||
| Nicotine + Sham (n = 5) | 126.4 ± 2.8 | 7.36 ± 0.4 | 34 ± 7.2 | 133 ± 37 | 135 ± 3 | 0 |
| 7.37 ± 0.04 | 38.0 ± 3.4 | 127 ± 28 | 135 ± 11.4 | |||
| Saline + tMCAO (n = 8) | 130.3 ± 8.7 | 7.36 ± 0.4 | 35.3 ± 2.3 | 134 ± 39 | 131 ± 7.7 | 1 |
| 7.37 ± 0.05 | 34.3 ± 5.2 | 140 ± 15 | 134 ± 9.3 | |||
| Nicotine + tMCAO (n = 8) | 127.4 ± 6.8 | 7.4 ± 0.2 | 38.08±4.5 | 130 ± 35 | 129 ± 7.8 | 2 |
| 7.39 ± 0.04 | 35.8 ± 3.9 | 138 ± 25 | 132 ± 5.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Adesky, N.D.; De Rivero Vaccari, J.P.; Bhattacharya, P.; Schatz, M.; Perez-Pinzon, M.A.; Bramlett, H.M.; Raval, A.P. Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats. Int. J. Mol. Sci. 2018, 19, 1330. https://doi.org/10.3390/ijms19051330
D’Adesky ND, De Rivero Vaccari JP, Bhattacharya P, Schatz M, Perez-Pinzon MA, Bramlett HM, Raval AP. Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats. International Journal of Molecular Sciences. 2018; 19(5):1330. https://doi.org/10.3390/ijms19051330
Chicago/Turabian StyleD’Adesky, Nathan D., Juan Pablo De Rivero Vaccari, Pallab Bhattacharya, Marc Schatz, Miguel A. Perez-Pinzon, Helen M. Bramlett, and Ami P. Raval. 2018. "Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats" International Journal of Molecular Sciences 19, no. 5: 1330. https://doi.org/10.3390/ijms19051330
APA StyleD’Adesky, N. D., De Rivero Vaccari, J. P., Bhattacharya, P., Schatz, M., Perez-Pinzon, M. A., Bramlett, H. M., & Raval, A. P. (2018). Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats. International Journal of Molecular Sciences, 19(5), 1330. https://doi.org/10.3390/ijms19051330