B-Myb Mediates Proliferation and Migration of Non-Small-Cell Lung Cancer via Suppressing IGFBP3

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Prognostic Significance of B-Myb in NSCLC (Non-Small-Cell Lung Cancer)
2.2. B-Myb Depletion Delays the Cell Cycle Progression and Inhibits Proliferation in Adenocarcinoma Cells (ADC)
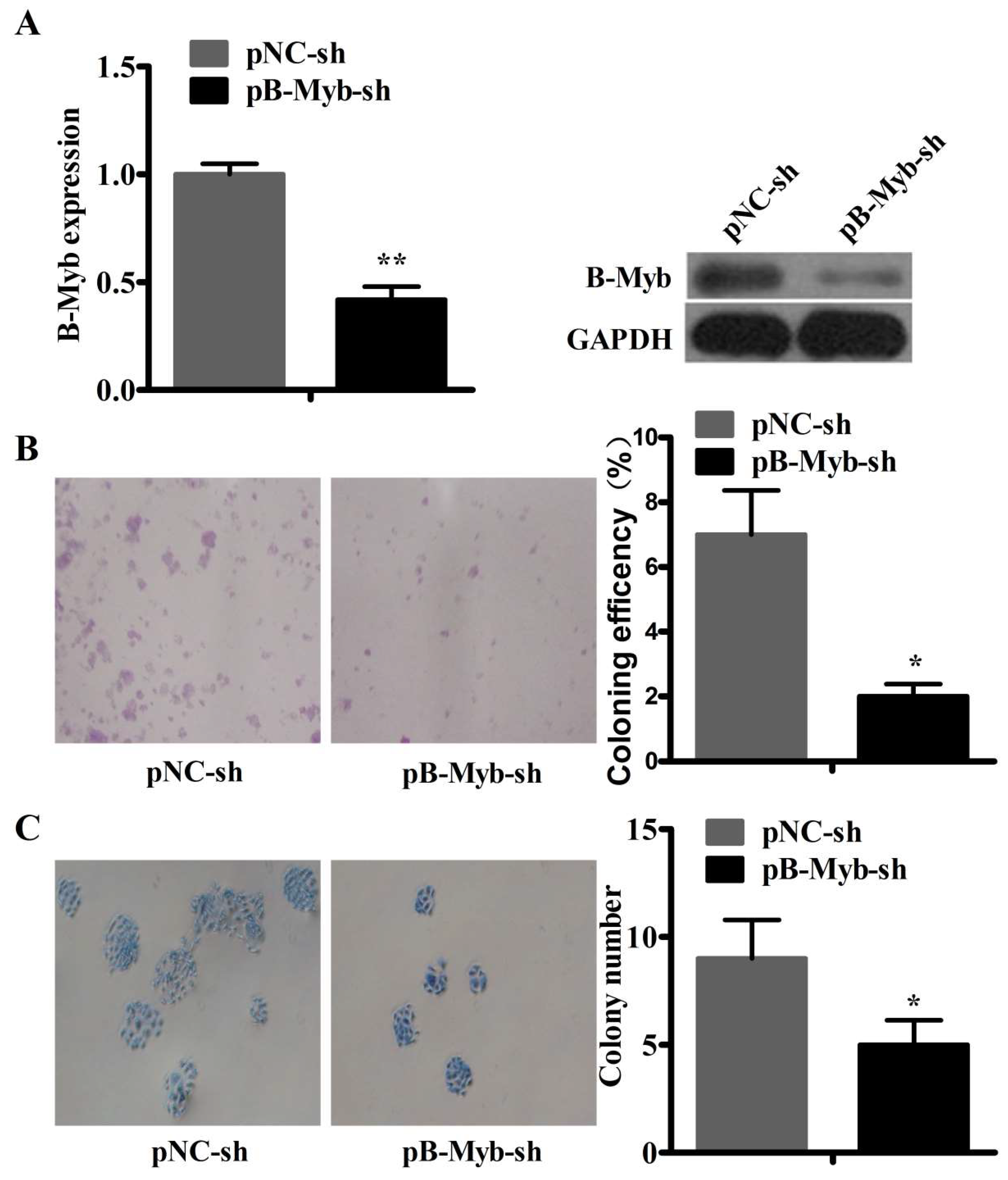
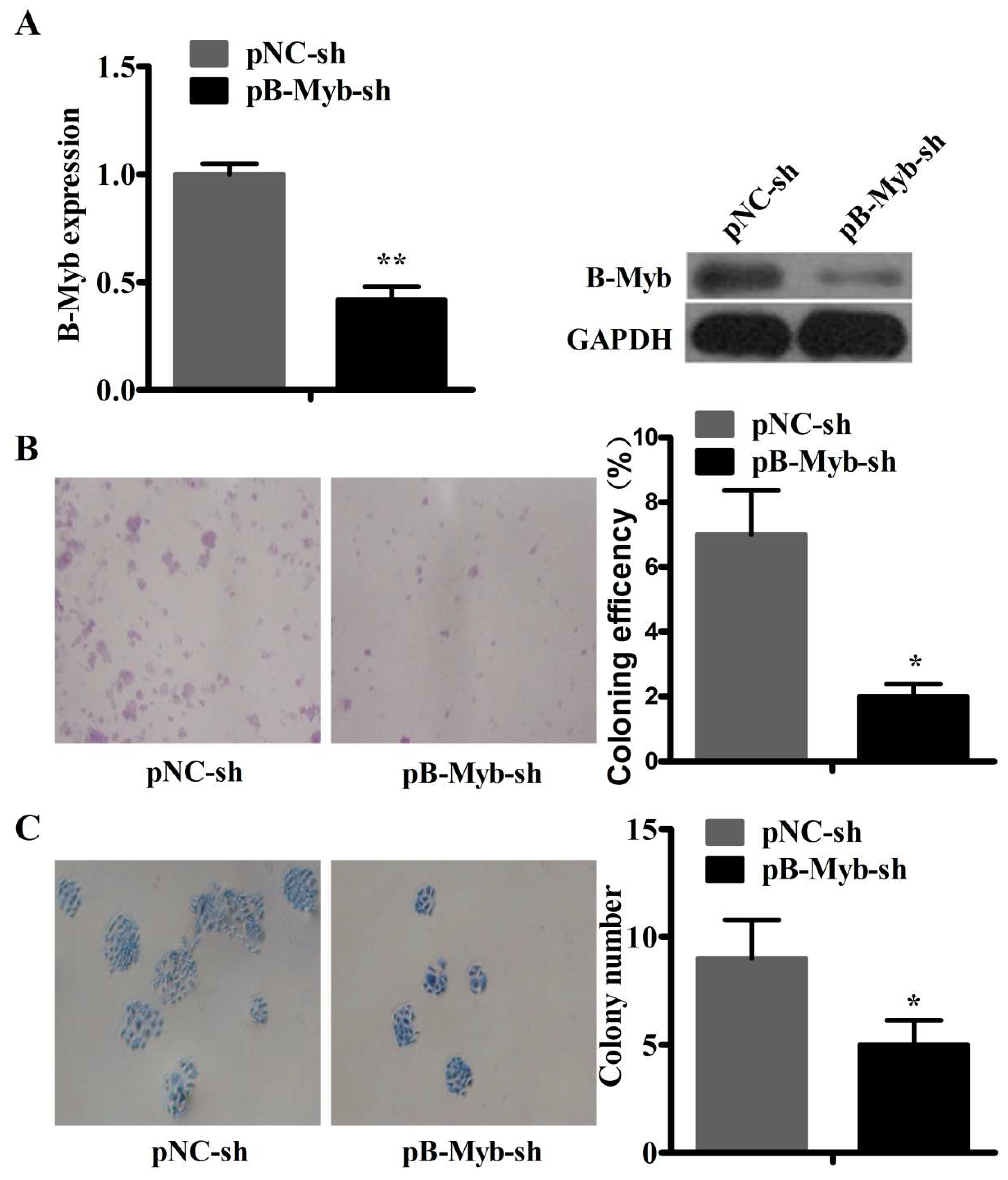
2.3. B-Myb Depletion Reduces Motility in A549 Lung Cancer Cells
2.4. B-Myb Depletion Inhibits Lung Tumorigenesis in Vitro and in Vivo
2.5. RNA-Seq Analysis after siRNA-Mediated B-Myb Knockdown
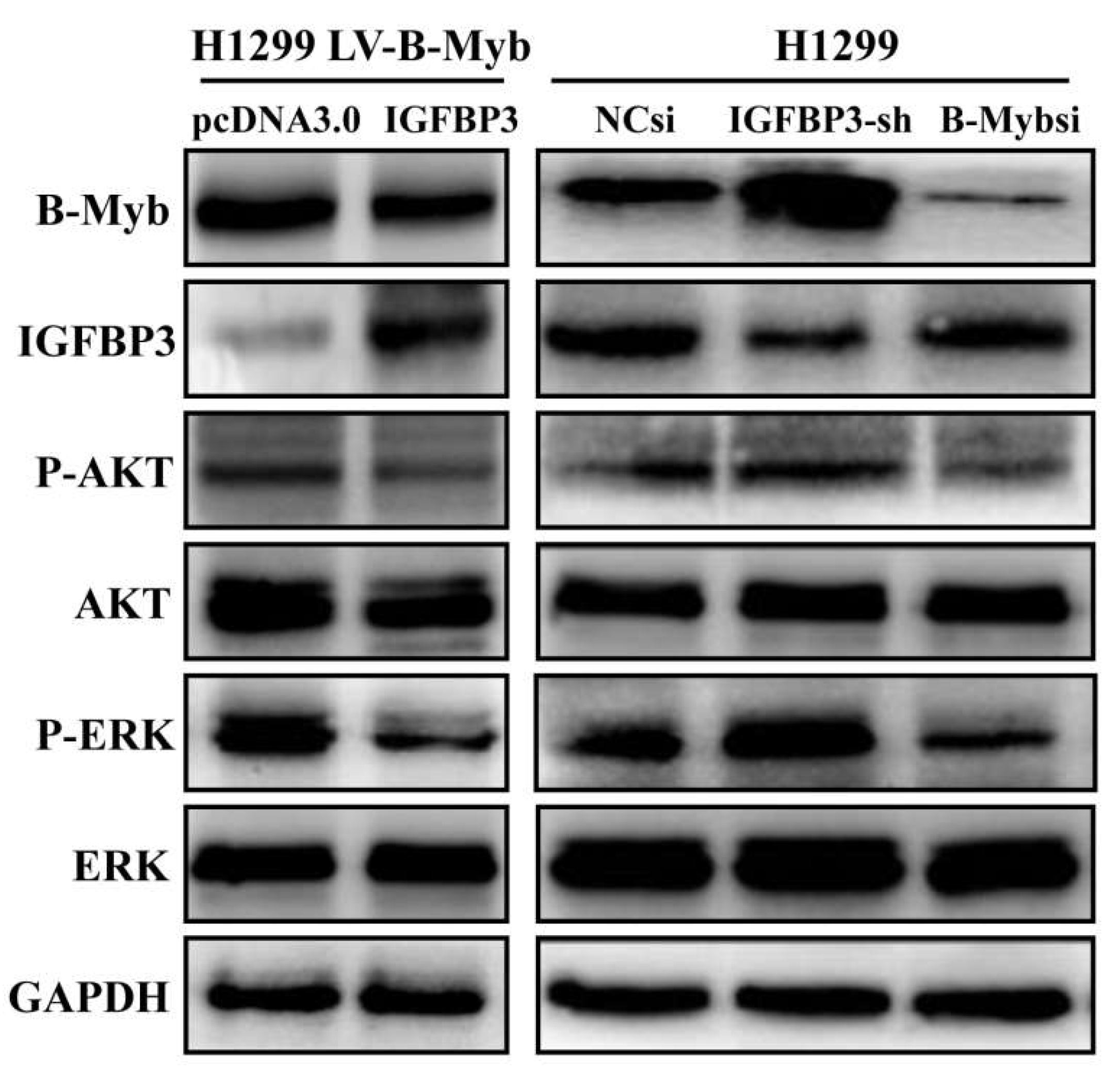
2.6. B-Myb Promotes Cell Proliferation and Migration by Targeting IGFBP3 in NSCLC Cells
2.7. B-Myb Activates ERK and Akt Pathways via Targeting IGFBP3 in NSCLC Cells
2.8. B-Myb Inhibits IGFBP3 Promoter Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. siRNA Synthesis and Transfection
4.3. RNA Isolation and qRT-PCR
4.4. Western Blot Analysis
4.5. Cell Proliferation Assay
4.6. Cell Cycle Assay
4.7. Colony-Formation Assay
4.8. Wound Healing Assay
4.9. Transwell Cell Migration Assay
4.10. Transfection
4.11. Luciferase Reporter Constructs and Reporter Assays
4.12. Lentiviral Infection and Construction of Stably Silencing B-Myb Gene A549 Cell Lines
4.13. Tumor Xenografts
4.14. RNA-Seq Analysis
4.15. Prognostic Analysis of B-Myb in Lung Cancer Patients
4.16. Lentivirus-Mediated Establishment of Stable B-Myb Overexpression Cell Line
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| NSCLC | non-small-cell lung cancer |
| Mybl2 | MYB proto-oncogene-like 2 |
| COL11A1 | collagen type XI alpha 1 chain |
| FLT4 | fms-related tyrosine kinase 4 |
| SPARC | secreted protein acidic and cysteine rich |
| IDH2 | isocitrate dehydrogenase (NADP(+)) 2 |
| IGFBP3 | insulin-like growth factor binding protein 3 |
| PDK3 | pyruvate dehydrogenase kinase 3 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.H.; Reddy, E.P. The Myb gene family in cell growth, differentiation and apoptosis. Oncogene 1999, 18, 3017–3033. [Google Scholar] [CrossRef] [PubMed]
- Joaquin, M.; Watson, R.J. Cell cycle regulation by the B-Myb transcription factor. Cell. Mol. Life Sci. 2003, 60, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Dimaio, D. B-Myb, cancer, senescence, and microRNAs. Cancer Res. 2011, 71, 5370–5373. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.; Bennett, J.D.; Watson, R.J. Cell-cycle regulation of human B-myb transcription. Gene 1995, 160, 277–281. [Google Scholar] [CrossRef]
- Sala, A.; Kundu, M.; Casella, I.; Engelhard, A.; Calabretta, B.; Grasso, L.; Paggi, M.G.; Giordano, A.; Watson, R.J.; Khalili, K.; et al. Activation of human B-MYB by cyclins. Proc. Natl. Acad. Sci. USA 1997, 94, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Sala, A. B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur. J. Cancer 2005, 41, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Kowenz-Leutz, E.; Dechend, R.; Leutz, A. B-Myb, a repressed trans-activating protein. J. Mol. Med. 1997, 75, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Raschella, G.; Negroni, A.; Sala, A.; Pucci, S.; Romeo, A.; Calabretta, B. Requirement of B-Myb function for survival and differentiative potential of human neuroblastoma cells. J. Biol. Chem. 1995, 270, 8540–8545. [Google Scholar] [CrossRef] [PubMed]
- Ahlbory, D.; Appl, H.; Lang, D.; Klempnauer, K.H. Disruption of B-Myb in DT40 cells reveals novel function for B-Myb in the response to DNA-damage. Oncogene 2005, 24, 7127–7134. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shira, A.; Pinthus, J.H.; Rozovsky, U.; Goldstein, M.; Sellers, W.R.; Yaron, Y.; Eshhar, Z.; Orr-Urtreger, A. Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res. 2002, 62, 6803–6807. [Google Scholar] [PubMed]
- Ren, F.; Wang, L.; Shen, X.; Xiao, X.; Liu, Z.; Wei, P.; Wang, Y.; Qi, P.; Shen, C.; Sheng, W.; et al. MYBL2 is an independent prognostic marker that has tumor-promoting functions in colorectal cancer. Am. J. Cancer Res. 2015, 5, 1542–1552. [Google Scholar] [PubMed]
- Calvisi, D.F.; Simile, M.M.; Ladu, S.; Frau, M.; Evert, M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.; Seddaiu, M.A.; Brozzetti, S.; et al. Activation of v-Myb avian myeloblastosis viral oncogene homolog-like2 (MYBL2)-LIN9 complex contributes to human hepatocarcinogenesis and identifies a subset of hepatocellular carcinoma with mutant p53. Hepatology 2011, 53, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Thorner, A.R.; Hoadley, K.A.; Parker, J.S.; Winkel, S.; Millikan, R.C.; Perou, C.M. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene 2009, 28, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, W.C.; Weiss, W.A. Addiction to B-MYB. Oncotarget 2010, 1, 235–236. [Google Scholar] [PubMed]
- Raschella, G.; Cesi, V.; Amendola, R.; Negroni, A.; Tanno, B.; Altavista, P.; Tonini, G.P.; de Bernardi, B.; Calabretta, B. Expression of B-myb in neuroblastoma tumors is a poor prognostic factor independent from MYCN amplification. Cancer Res. 1999, 59, 3365–3368. [Google Scholar] [PubMed]
- Fuster, O.; Llop, M.; Dolz, S.; Garcia, P.; Such, E.; Ibanez, M.; Luna, I.; Gomez, I.; Lopez, M.; Cervera, J.; et al. Adverse prognostic value of MYBL2 overexpression and association with microRNA-30 family in acute myeloid leukemia patients. Leuk. Res. 2013, 37, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhu, H.; Cai, W.; Fan, X.; Wang, Y.; Niu, Y.; Song, F.; Bu, Y. B-Myb is Up-regulated and promotes cell growth and motility in non-small cell lung cancer. Int. J. Mol. Sci. 2017, 18, 860. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.J.; Firth, S.M.; King, M.A.; Baxter, R.C. Insulin-like growth factor-binding protein-3 modulates expression of Bax and Bcl-2 and potentiates p53-independent radiation-induced apoptosis in human breast cancer cells. J. Biol. Chem. 2000, 275, 39174–39181. [Google Scholar] [CrossRef] [PubMed]
- Rajah, R.; Valentinis, B.; Cohen, P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-β1 on programmed cell death through a p53- and IGF-independent mechanism. J. Biol. Chem. 1997, 272, 12181–12188. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Chun, K.H.; Liu, B.; Wiehle, S.A.; Cristiano, R.J.; Hong, W.K.; Cohen, P.; Kurie, J.M. Insulin-like growth factor binding protein-3 inhibits the growth of non-small cell lung cancer. Cancer Res. 2002, 62, 3530–3537. [Google Scholar] [PubMed]
- Wang, H.H.; Wang, Y.C.; Wu, D.W.; Hung, C.S.; Chen, C.Y.; Lee, H. Targeting insulin-like growth factor-binding protein-3 by microRNA-125b promotes tumor invasion and poor outcomes in non-small-cell lung cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; dePrimo, S.E.; Grabert, L.M.; Fu, V.X.; Brooks, J.D.; Jarrard, D.F. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J. Biol. Chem. 2002, 277, 14877–14883. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Moerman, E.J.; Jones, R.A.; Baxter, R.C. Insulin-like growth factor binding protein 3 accumulates to high levels in culture medium of senescent and quiescent human fibroblasts. Proc. Natl. Acad. Sci. USA 1991, 88, 9680–9684. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A.; Cohen, P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J. Cell. Physiol. 2000, 183, 1–9. [Google Scholar] [CrossRef]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forozan, F.; Mahlamaki, E.H.; Monni, O.; Chen, Y.; Veldman, R.; Jiang, Y.; Gooden, G.C.; Ethier, S.P.; Kallioniemi, A.; Kallioniemi, O.P. Comparative genomic hybridization analysis of 38 breast cancer cell lines: A basis for interpreting complementary DNA microarray data. Cancer Res. 2000, 60, 4519–4525. [Google Scholar] [PubMed]
- Nakajima, T.; Yasui, K.; Zen, K.; Inagaki, Y.; Fujii, H.; Minami, M.; Tanaka, S.; Taniwaki, M.; Itoh, Y.; Arii, S.; et al. Activation of B-Myb by E2F1 in hepatocellular carcinoma. Hepatol. Res. 2008, 38, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Pan, Y.; Lu, H.; Zheng, S.; Lin, H.; Fang, H.; Cao, F. B-myb is a gene implicated in cell cycle and proliferation of breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 5819–5827. [Google Scholar] [PubMed]
- Mowla, S.N.; Lam, E.W.; Jat, P.S. Cellular senescence and aging: The role of B-MYB. Aging Cell 2014, 13, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Cervera, N.; Finetti, P.; Esteyries, S.; Esterni, B.; Adelaide, J.; Xerri, L.; Viens, P.; Jacquemier, J.; Charafe-Jauffret, E.; et al. Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin. Cancer Res. 2006, 12, 4533–4544. [Google Scholar] [CrossRef] [PubMed]
- Jazaeri, A.A.; Lu, K.; Schmandt, R.; Harris, C.P.; Rao, P.H.; Sotiriou, C.; Chandramouli, G.V.; Gershenson, D.M.; Liu, E.T. Molecular determinants of tumor differentiation in papillary serous ovarian carcinoma. Mol. Carcinog. 2003, 36, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tang, L. SPARC in Tumor Pathophysiology and as a Potential Therapeutic Target. Curr. Pharm. Des. 2014, 20, 6182–6190. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Dontula, R.; El-Rayes, B.F.; Lakka, S.S. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis 2014, 35, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Sinn, B.V.; Striefler, J.K.; Lindner, J.L.; Stieler, J.M.; Lohneis, P.; Bischoff, S.; Blaker, H.; Pelzer, U.; Bahra, M.; et al. SPARC expression in resected pancreatic cancer patients treated with gemcitabine: Results from the CONKO-001 study. Ann. Oncol. 2014, 25, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; La Cava, P.; Tibullo, D.; Barbagallo, I.; Parrinello, N.; Cupri, A.; Stagno, F.; Consoli, C.; Chiarenza, A.; Palumbo, G.A.; et al. SPARC expression in CML is associated to imatinib treatment and to inhibition of leukemia cell proliferation. BMC Cancer 2013, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Li, Y.Y.; Shao, Q.; Hou, J.H.; Wang, F.; Cai, M.B.; Zeng, Y.X.; Shao, J.Y. Secreted protein acidic and rich in cysteine (SPARC) is associated with nasopharyngeal carcinoma metastasis and poor prognosis. J. Transl. Med. 2012, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, W.; Wang, C.; Liu, F.; Guan, S.; Sun, Y.; Wang, X.; An, D.; Wen, Z.; Chen, P.; et al. The clinical significance of isocitrate dehydrogenase 2 in esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 700–714. [Google Scholar] [PubMed]
- Lv, Q.; Xing, S.; Li, Z.; Li, J.; Gong, P.; Xu, X.; Chang, L.; Jin, X.; Gao, F.; Li, W.; et al. Altered expression levels of IDH2 are involved in the development of colon cancer. Exp. Ther. Med. 2012, 4, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Hung, C.L.; Chen, Y.R.; Yang, J.C.; Wang, J.; Campbell, M.; Izumiya, Y.; Chen, H.W.; Wang, W.C.; Ann, D.K.; et al. KDM4A coactivates E2F1 to regulate the PDK-dependent metabolic switch between mitochondrial oxidation and glycolysis. Cell Rep. 2016, 16, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, M.A. Phosphatidylinositol-3 kinase dependent pathways: The role in control of cell growth, survival, and malignant transformation. Biochem. Biokhimiia 2000, 65, 59–67. [Google Scholar]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Kulik, G.; Weber, M.J. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol. Cell. Biol. 1998, 18, 6711–6718. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Su, Y.H.; Chuang, R.L.; Chang, T.Y. Suppression of transforming growth factor-β-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene 1998, 17, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M.; Smith, C.E.; Mashiba, M.K.; Okawa, T.; Andl, C.D.; El-Deiry, W.S.; Nakagawa, H. EGF-mediated regulation of IGFBP-3 determines esophageal epithelial cellular response to IGF-I. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G404–G416. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Wuputra, K.; Liu, C.J.; Lin, Y.C.; Chen, Y.T.; Chai, C.Y.; Lin, C.S.; Kuo, K.K.; Tsai, M.H.; Wang, S.W.; et al. Oncogenic function of the homeobox A13-long noncoding RNA HOTTIP-insulin growth factor-binding protein 3 axis in human gastric cancer. Oncotarget 2016, 7, 36049–36064. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Multivariate Analysis | Univariate Analysis | ||
|---|---|---|---|---|
| HR (95% CI) a | p Value b | HR (95% CI) a | p Value b | |
| 117 lung cancer patients | ||||
| B-Myb (High/Low) | 1.789 (0.974–3.286) | 0.043 | 1.870 (1.024–3.416) | 0.042 |
| AGE (≥60/<60) | 1.145 (0.627–2.090) | 0.660 | ||
| SEX (Male/Female) | 1.456 (0.808–2.623) | 0.211 | ||
| T (T1, T2/T3, T4) | 1.785 (0.82–3.886) | 0.144 | ||
| N (N0/N1, N2) | 2.533 (1.403–4.573) | 0.002 | 2.413 (1.347–4.326) | 0.003 |
| Pathway | Counts | Dysregulated Genes | p Value |
|---|---|---|---|
| MAPK signaling pathway | 59 | ACVR1C, CASP1, CASP14, CASP5, CD14, COL11A1, DDIT3, DUSP10, EGFR, ELK4, FAS, FASLG, FGF1, FGF10, FGF11, FGF13, FGF17, FGF19, FGF22, FGF8, FGFR4, FLT4, FOS, GADD45A, GNA12, HSPA9B, IL1A, IL1B, IL1R1, JUN, MAP2K5, MAP2K6, MAP3K14, MAP3K8, MAPK11, MAPK6, MAPK8IP2, MRAS, NFATC2, NTF3, NTRK1, PDGFRA, PLA2G10, PLA2G4A, PRKCB1, PRKX, PRKY, PTPN7, PTPRR, RAP1A, RASGRF1, RASGRP3, RASGRP4, RPS6KA4, STMN1, TGFB2, TMEM37, TNF, TP53 | 3.2 × 10−3 |
| Cytokine-cytokine receptor interaction | 83 | TNFSF15, TNFSF4, TNFRSF4, MPL, TNF, HGF, CCL27, TNFSF12, CCL2, TNFRSF8, TNFSF10, TNFSF8, TNFSF9, IFNLR1, TNFSF14, TNFRSF6B, CCL5, KITLG, TNFRSF19, IFNA1, CXCR4, TNFRSF17, CXCR2, IL7R, IL12RB, IL12B, IL13, IL23A, IL11RA, IL12A, CTF1, FLT1, FLT3, IL1A, FLT4, TNFRSF1B, CD27, TNFRSF12A, CXCR6, CCL20, EDAR, CCL21, IFNA13, IL10, TNFRSF13C, CSF1R, LTA, CSF1, IL3RA, CNTF, EGFR, IL2RG, IL2RB, TNFRSF9, OSM, IL15RA, IFNE, CSF2RA, TGFB2, CXCL13, LTB, CXCL11, CCR9, NGFR, IL1B, AMHR2, IL1R1, AMH, IL20RB, TNFRSF25, IL22RA1, LTBR, CXCR3, IL20RA, PDGFRA, GHR, CCR10, FAS, LIF, FASLG, IFNW1, IL4, TSLP | 6.39 × 10−6 |
| Transcriptional regulation in cancer | 57 | PAX8, HIST1H3F, ELK4, ZBTB16, ITGAM, CEBPB, TCF3, FCGR1A, SUPT3H, IGFBP3, PLAT, SIX1, MMP, 3 HIST2H3C, FLT1, GRIA3, SPI1, ID2, FLT3, MDM2, TFE3, UTY, SPINT1, CD14, CDK14, HIST1H3G, HIST1H3E, HIST1H3C, HIST3H3, HIST2H3A, HIST1H3J, CSF1R, TSPAN7, PBX1, IL2RB, RUNX1T1, ERG, TAF15, CDKN2C, ELANE, DDIT3, ETV7, NGFR, REL, BCL2A1, TMPRSS2, TP53, ATF1, HIST2H3D, NUPR1, EYA1, HDAC, 1 SLC45A3, CCNT2, NTRK1, LMO2, SSX1 | 1.24 × 10−3 |
| cAMP signaling pathway | 57 | JUN, ADCY8, CALM2, ADCY7, NPY, PTGER3, CALM1, HCAR3, GRIA4, ATP1A4, ATP1B1, PPP1R12A, GRIN1, GRIN2A, GRIN2B, CACNA1D, CFTR, VAV1, TNNI3, GRIA3, CACNA1F, GRIA1, ADCY4, ATP1A3, ATP1A2, HTR1B, CREB5, TIAM1, HCAR2, PIK3R5, ADORA2A, PDE3B, PDE4A, SSTR1, PDE4D, HTR6, CHRM2, ADCY2, ADCY1, PDE3A, AMH, NPR, 1 RAP1A, GLI1, PIK3R3, CAMK2A, CAMK2D, HCAR1, GLI3, PTCH1, VIPR2, HCN4, ACOX1, CNGA1, CALM3, FOS, GRIN3A | 0.045166 |
| Rap1 signaling pathway | 60 | ADCY8, ANGPT4, CALM2, ADCY7, PRKCB, SIPA1L1, CALM, 1 HGF, FGF8, ANGPT2, ITGAM, PDGFD, KITLG, FGF1, F2RL3, RAPGEF2, GRIN1, GRIN2A, GRIN2B, MAPK11, ID1, FLT1, FLT4, MAP2K6, FGFR4, ADCY4, LPAR5, EFNA2, P2RY1, FGF17, FGF10, RASGRP3, ITGB2, FGF11, SKAP1, TIAM1, FGF13, FGF19, PIK3R5, ADORA2A, CSF1R, CSF1, FPR1, EGFR, LAT, RASSF5, ADCY2, ADCY1, MRAS, NGFR, CNR1, RAP1A, PIK3R3, FGF22, RALB, PDGFRA, TEK, CALM3, FYB, CDH1 | 0.045802 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Wang, Y.; Jiang, T.; Cai, W.; Jin, Y.; Niu, Y.; Zhu, H.; Bu, Y. B-Myb Mediates Proliferation and Migration of Non-Small-Cell Lung Cancer via Suppressing IGFBP3. Int. J. Mol. Sci. 2018, 19, 1479. https://doi.org/10.3390/ijms19051479
Fan X, Wang Y, Jiang T, Cai W, Jin Y, Niu Y, Zhu H, Bu Y. B-Myb Mediates Proliferation and Migration of Non-Small-Cell Lung Cancer via Suppressing IGFBP3. International Journal of Molecular Sciences. 2018; 19(5):1479. https://doi.org/10.3390/ijms19051479
Chicago/Turabian StyleFan, Xiaoyan, Yitao Wang, Tinghui Jiang, Wei Cai, Yuelei Jin, Yulong Niu, Huifang Zhu, and Youquan Bu. 2018. "B-Myb Mediates Proliferation and Migration of Non-Small-Cell Lung Cancer via Suppressing IGFBP3" International Journal of Molecular Sciences 19, no. 5: 1479. https://doi.org/10.3390/ijms19051479