New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review

 and
and
Abstract
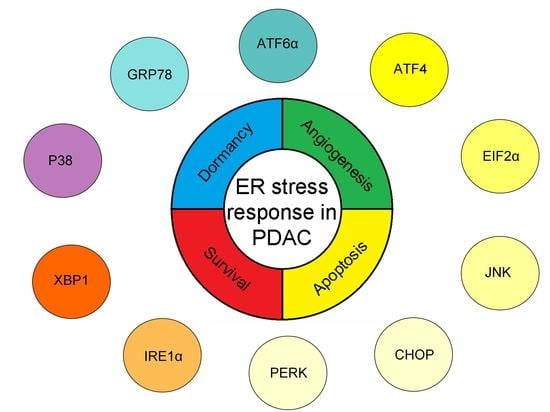
:
1. Introduction
2. Endoplasmic Reticulum Stress Response in PDAC
3. ERSR to Induce Dormancy in PDAC
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF6α | activating transcription factor 6 isoform α |
| AKT | AKT serine/threonine kinase |
| ATM | Ataxia telangiectasia mutated |
| CHOP | CCAAT/enhancer binding protein (C/EBP)-homologous protein |
| CI | confident interval |
| CSC | cancer stem cells |
| GRP78 | 78 kDa glucose-regulated protein |
| CT | computed tomography |
| eIF2α | eukaryotic initiation factor 2 isoform α |
| EMT | epithelial-mesenchymal transition |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| ERSR | endoplasmic reticulum stress response |
| FAP | Familial adenomatous polyposis |
| HR | hazard ratio |
| IRE1 | inositol-requiring enzyme 1 |
| JNK | c-Jun N-terminal kinase |
| MAPKs | mitogen-activated protein kinases |
| MRI | magnetic resonance imaging |
| NCCN | National Comprehensive Cancer Network |
| OS | overall survival |
| PARP | poly ADP ribose polymerase |
| PDAC | pancreatic ductal adenocarcinoma |
| PERK | protein kinase RNA-like endoplasmic reticulum kinase |
| PJS | Peutz-Jeghers Syndrome |
| UP | unfolded protein |
| UPR | unfolded protein response |
| VEGF | vascular endothelial growth factor |
| XBP1 | transcription factor X-box 1 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Bertuccio, P.; Negri, E.; La Vecchia, C.; Zeegers, M.P.; Boffetta, P. Pancreatic cancer: Overview of descriptive epidemiology. Mol. Carcinog. 2012, 51, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pancreatic Cancer Action Network. The Alarming Rise of Pancreatic Cancer Deaths in the United States: Why We Need to Stem the Tide Today. 2016. Available online: https://www.pancan.org/wp-content/uploads/2013/01/incidence_report_2012_executive_summary.pdf (accessed on 16 February 2016).
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Consumption of sugar and sugar-sweetened foods and the risk of pancreatic cancer in a prospective study. Am. J. Clin. Nutr. 2006, 84, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B. Risk factors for pancreatic cancer: A summary review of meta-analytical studies. Int. J. Epidemiol. 2015, 44, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Yeo, T.P. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin. Oncol. 2015, 42, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Red and processed meat consumption and risk of pancreatic cancer: Meta-analysis of prospective studies. Br. J. Cancer 2012, 106, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Ghadirian, P.; Baillargeon, J.; Simard, A.; Perret, C. Food habits and pancreatic cancer: A case-control study of the francophone community in Montreal, Canada. Cancer Epidemiol. Biomark. Prev. 1995, 4, 895–899. [Google Scholar]
- Amaral, A.F.; Porta, M.; Silverman, D.T.; Milne, R.L.; Kogevinas, M.; Rothman, N.; Cantor, K.P.; Jackson, B.P.; Pumarega, J.A.; Lopez, T.; et al. Pancreatic cancer risk and levels of trace elements. Gut 2012, 61, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- WHO Guidelines. Water Quality and Health Strategy 2013–2020. Available online: http://www.who.int/water_sanitation_health/dwq/water_quality_strategy.pdf?ua=1 (accessed on 4 July 2013).
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Klein, A.P. Genome-wide sequencing to identify the cause of hereditary cancer syndromes: With examples from familial pancreatic cancer. Cancer Lett. 2013, 340, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P. Genetic predisposition to pancreatic cancer. World J. Gastroenterol. 2014, 20, 10778–10789. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, H.; Takaori, K.; Morizane, C.; Maguchi, H.; Mizuma, M.; Takahashi, H.; Wada, K.; Hosoi, H.; Yachida, S.; Suzuki, M.; et al. Familial pancreatic cancer: Concept, management and issues. World J. Gastroenterol. 2017, 23, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Offerhaus, G.J.; Lee, D.H.; Krush, A.J.; Tersmette, A.C.; Booker, S.V.; Kelley, N.C.; Hamilton, S.R. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut 1993, 34, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Korsse, S.E.; Harinck, F.; van Lier, M.G.; Biermann, K.; Offerhaus, G.J.; Krak, N.; Looman, C.W.; van Veelen, W.; Kuipers, E.J.; Wagner, A.; et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: A large cohort study and implications for surveillance. J. Med. Genet. 2013, 50, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of pancreatic cancer in families with lynch syndrome. JAMA 2009, 302, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Garcia-Foncillas, J. Can molecular biomarkers change the paradigm of pancreatic cancer prognosis? Biomed. Res. Int. 2016, 2016, 4873089. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Garcia-Foncillas, J. The role of BRCA2 mutation status as diagnostic, predictive, and prognosis biomarker for pancreatic cancer. Biomed. Res. Int. 2016, 2016, 1869304. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, S.; Nakamura, Y.; Atsumi, Y.; Murakawa, M.; Yamaoku, K.; Aoyama, T.; Kobayashi, S.; Ueno, M.; Morimoto, M.; Yokose, T.; et al. Locked nucleic acid in situ hybridization analysis of MicroRNA-21 predicts clinical outcome in patients after resection for pancreatic cancer treated with adjuvant gemcitabine monotherapy. Anticancer Res. 2016, 36, 1083–1088. [Google Scholar] [PubMed]
- Wei, X.; Wang, W.; Wang, L.; Zhang, Y.; Zhang, X.; Chen, M.; Wang, F.; Yu, J.; Ma, Y.; Sun, G. MicroRNA-21 induces 5-fluorouracil resistance in human pancreatic cancer cells by regulating PTEN and PDCD4. Cancer Med. 2016, 5, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Cunningham, D.; Peckitt, C.; Barton, S.; Tait, D.; Hawkins, M.; Watkins, D.; Starling, N.; Rao, S.; Begum, R.; et al. miR-21 expression and clinical outcome in locally advanced pancreatic cancer: Exploratory analysis of the pancreatic cancer Erbitux, radiotherapy and UFT (PERU) trial. Oncotarget 2016, 7, 12672–12681. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.P.; Burke, W.; Chen, R.; Bennett, R.L.; Schmidt, R.A.; Dellinger, E.P.; Kimmey, M.; Crispin, D.; Brentnall, T.A.; Byrd, D.R. Familial pancreatic adenocarcinoma: Association with diabetes and early molecular diagnosis. J. Med. Genet. 1995, 32, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Kenner, B.J.; Chari, S.T.; Maitra, A.; Srivastava, S.; Cleeter, D.F.; Go, V.L.; Rothschild, L.J.; Goldberg, A.E. Early detection of pancreatic cancer, a defined future using lessons from other cancers: A white paper. Pancreas 2016, 45, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Egawa, S.; Takeda, K.; Fukuyama, S.; Motoi, F.; Sunamura, M.; Matsuno, S. Clinicopathological aspects of small pancreatic cancer. Pancreas 2004, 28, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ariyama, J.; Suyama, M.; Satoh, K.; Sai, J. Imaging of small pancreatic ductal adenocarcinoma. Pancreas 1998, 16, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Redaelli, C.; Lietz, M.; Seiler, C.A.; Friess, H.; Buchler, M.W. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br. J. Surg. 2004, 91, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Cucchetti, A.; Ercolani, G.; Taffurelli, G.; Serenari, M.; Maroni, L.; Pezzilli, R.; Del Gaudio, M.; Ravaioli, M.; Cescon, M.; Pinna, A.D. A comprehensive analysis on expected years of life lost due to pancreatic cancer. Pancreatology 2016, 16, 449–453. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. NCCN Guidelines Pancreatic Adenocarcinoma. Version 1. 2017. Available online: https://www.nccn.org/patients/guidelines/pancreatic/index.html (accessed on 3 March 2017).
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Guidelines. Pathology and Genetics of Tumours of the Digestive System. In World Health Organization Classification of Tumours, 4th ed.; IARC: Lyon, France, 2010; Volume 3. [Google Scholar]
- Burris, H.A., III; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Bird, N.T.; Elmasry, M.; Jones, R.; Psarelli, E.; Dodd, J.; Malik, H.; Greenhalf, W.; Kitteringham, N.; Ghaneh, P.; Neoptolemos, J.P.; et al. Immunohistochemical hent1 expression as a prognostic biomarker in patients with resected pancreatic ductal adenocarcinoma undergoing adjuvant gemcitabine-based chemotherapy. Br. J. Surg. 2017, 104, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Hurt, C.N.; Bridgewater, J.; Falk, S.; Cummins, S.; Wasan, H.; Crosby, T.; Jephcott, C.; Roy, R.; Radhakrishna, G.; et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): A multicentre, randomised, phase 2 trial. Lancet Oncol. 2013, 14, 317–326. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Goldstein, D.; Renschler, M.F. Albumin-bound paclitaxel plus gemcitabine in pancreatic cancer. N. Engl. J. Med. 2014, 370, 479–480. [Google Scholar] [PubMed]
- O’Reilly, E.M.; Lowery, M.A.; Segal, M.F.; Smith, S.C.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; Salo-Mullen, E.E.; Hollywood, E.; et al. Phase IB trial of cisplatin (C), gemcitabine (G), and veliparib (V) in patients with known or potential BRCA or PALB2-mutated pancreas adenocarcinoma (PC). J. Clin. Oncol. 2014, 32, 4023. [Google Scholar]
- Fogelman, D.R.; Wolff, R.A.; Kopetz, S.; Javle, M.; Bradley, C.; Mok, I.; Cabanillas, F.; Abbruzzese, J.L. Evidence for the efficacy of iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res. 2011, 31, 1417–1420. [Google Scholar] [PubMed]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H., III. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the cancer and leukemia group b (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- ESMO Guidelines Committee. Eupdate—Cancer of the Pancreas Treatment Recommendations. 2017. Available online: www.esmo.org/Guidelines/Gastrointestinal-Cancers/Cancer-of-the-Pancreas/eUpdate-Treatment-Recommendations (accessed on 20 June 2017).
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, P.E.; Chambers, A.F. Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer 2010, 10, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell. Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, R.J. Molecular chaperones and the heat shock response. Sponsored by cold spring harbor laboratory, 6–10 May 1998. Biochim. Biophys. Acta 1999, 1423, R13–R27. [Google Scholar] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, Z.Q.; Zhang, K. Endoplasmic reticulum stress response in cancer: Molecular mechanism and therapeutic potential. Am. J. Transl. Res. 2010, 2, 65–74. [Google Scholar] [PubMed]
- Namba, T.; Kodama, R. Avarol induces apoptosis in pancreatic ductal adenocarcinoma cells by activating PERK-eIF2á-CHOP signaling. Mar. Drugs 2015, 13, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Wang, M.; Zhou, L.; Yao, L.; Liao, Q.; Zhao, Y. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci. Rep. 2015, 5, 16067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.P.; Dong, M.; Li, X.; Zhou, J.P. GRP78 promotes the invasion of pancreatic cancer cells by FAK and JNK. Mol. Cell. Biochem. 2015, 398, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. GRP78-mediated antioxidant response and abc transporter activity confers chemoresistance to pancreatic cancer cells. Mol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Raiter, A.; Weiss, C.; Bechor, Z.; Ben-Dor, I.; Battler, A.; Kaplan, B.; Hardy, B. Activation of GRP78 on endothelial cell membranes by an ADAM15-derived peptide induces angiogenesis. J. Vasc. Res. 2010, 47, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Stapleton, C.; Luo, B.; Xiong, S.; Ye, W.; Zhang, Y.; Jhaveri, N.; Zhu, G.; Ye, R.; Liu, Z.; et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011, 71, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chen, Y.; Huang, S.; Cui, F.; Wang, X. Suppression of GRP78 sensitizes human colorectal cancer cells to oxaliplatin by downregulation of CD24. Oncol. Lett. 2018, 15, 9861–9867. [Google Scholar] [CrossRef] [PubMed]
- Kawiak, A.; Domachowska, A.; Jaworska, A.; Lojkowska, E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and BIK upregulation. Sci. Rep. 2017, 7, 43781. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.A.; Kapoor, V.; Hoye, K.; Khudanyan, A.; Collins, A.; Thotala, D.; Hallahan, D.E. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin. Cancer Res. 2017, 23, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.L.; Su, C.C. Tanshinone IIA increases protein expression levels of PERK, ATF6, IRE1á, CHOP, caspase3 and caspase12 in pancreatic cancer BxPC3 cell derived xenograft tumors. Mol. Med. Rep. 2017, 15, 3259–3263. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. Chop induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PLoS ONE 2009, 4, e8008. [Google Scholar] [CrossRef]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.R.; Singleton, D.C.; Buffa, F.; Abramczyk, O.; Phadwal, K.; Li, J.L.; Simon, A.K.; Murray, J.T.; Harris, A.L. Transcriptional up-regulation of ulk1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013, 449, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.H.; Ueda, E.; Bortner, C.D.; Yang, X.P.; Liao, G.; Jetten, A.M. Farnesol activates the intrinsic pathway of apoptosis and the ATF4-ATF3-chop cascade of ER stress in human t lymphoblastic leukemia molt4 cells. Biochem. Pharmacol. 2015, 97, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Yin, Y.; Liu, H.Y.; Feng, Y.Y.; Bian, Z.H.; Zhou, L.Y.; Zhang, J.W.; Fei, B.J.; Wang, Y.G.; Huang, Z.H. Glucose deprivation induces chemoresistance in colorectal cancer cells by increasing ATF4 expression. World J. Gastroenterol. 2016, 22, 6235–6245. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, P.; Xi, D.; Zhu, H.; Gao, Y. ATF4 regulates CCL2 expression to promote endometrial cancer growth by controlling macrophage infiltration. Exp. Cell Res. 2017, 360, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Klieser, E.; Illig, R.; Stattner, S.; Primavesi, F.; Jager, T.; Swierczynski, S.; Kiesslich, T.; Kemmerling, R.; Bollmann, C.; Di Fazio, P.; et al. Endoplasmic reticulum stress in pancreatic neuroendocrine tumors is linked to clinicopathological parameters and possible epigenetic regulations. Anticancer Res. 2015, 35, 6127–6136. [Google Scholar] [PubMed]
- Palam, L.R.; Gore, J.; Craven, K.E.; Wilson, J.L.; Korc, M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015, 6, e1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nami, B.; Donmez, H.; Kocak, N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24− phenotype breast cancer stem cells. Exp. Toxicol. Pathol. 2016, 68, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting xbp-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α -driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Samur, M.; Munshi, A.; Hideshima, T.; Keskin, D.; Kimmelman, A.; Lee, A.H.; Dranoff, G.; Anderson, K.C.; Munshi, N.C. Heteroclitic XBP1 peptides evoke tumor-specific memory cytotoxic t lymphocytes against breast cancer, colon cancer, and pancreatic cancer cells. Oncoimmunology 2014, 3, e970914. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksa, G.; Nikolos, F.; Bado, I.; Clarke, R.; Gustafsson, J.A.; Thomas, C. ERâ decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 2015, 34, 4130–4141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (er) stress and causes translocation of ATF6 from the ER to the golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Parmar, V.M.; Schroder, M. Sensing endoplasmic reticulum stress. Adv. Exp. Med. Biol. 2012, 738, 153–168. [Google Scholar] [PubMed]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, T.; Odisho, T.; Sidorova, E.; Volchuk, A. Pancreatic â- cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell Physiol. 2012, 302, C992–C1003. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Georgiev-Hristov, T.; Borrero-Palacios, A.; Fernandez-Acenero, M.J.; Rodriguez-Remirez, M.; del Puerto-Nevado, L.; Cebrian, A.; Gomez del Pulgar, M.T.; Cazorla, A.; Vega-Bravo, R.; et al. Identification of poor-outcome biliopancreatic carcinoma patients with two-marker signature based on ATF6α and p-p38 “Stard compliant”. Medicine 2015, 94, e1972. [Google Scholar] [CrossRef] [PubMed]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Tay, K.H.; Luan, Q.; Croft, A.; Jiang, C.C.; Jin, L.; Zhang, X.D.; Tseng, H.Y. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal. 2014, 26, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Shpigner, L.; Raanan, C.; Schreiber, L.; Melzer, E.; Seger, R. Dynamic distribution of ERK, p38 and JNK during the development of pancreatic ductal adenocarcinoma. Acta Histochem. 2014, 116, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Naito, Y.; Cope, L.; Naranjo-Suarez, S.; Saunders, T.; Hong, S.M.; Goggins, M.G.; Herman, J.M.; Wolfgang, C.L.; Iacobuzio-Donahue, C.A. Functional p38 MAPK identified by biomarker profiling of pancreatic cancer restrains growth through JNK inhibition and correlates with improved survival. Clin. Cancer Res. 2014, 20, 6200–6211. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Sharon, C.; Baranwal, S.; Boothello, R.S.; Desai, U.R.; Patel, B.B. Heparan sulfate hexasaccharide selectively inhibits cancer stem cells self-renewal by activating p38 map kinase. Oncotarget 2016, 7, 84608–84622. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The relationship between dormant cancer cells and their microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α -Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Q.; Feng, X.; Bo, Y. Effect of evodiagenine mediates photocytotoxicity on human breast cancer cells MDA-MB-231 through inhibition of PI3K/AKT/mTOR and activation of p38 pathways. Fitoterapia 2014, 99, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J., III; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ha, D.P.; Zhu, G.; Rangel, D.F.; Kobielak, A.; Gill, P.S.; Groshen, S.; Dubeau, L.; Lee, A.S. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant kras-driven pancreatic tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E4020–E4029. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Lulla, A.R.; Wagner, J.; Ralff, M.D.; Kiehl, J.B.; Zhou, Y.; Benes, C.H.; Prabhu, V.V.; Oster, W.; Astsaturov, I.; et al. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF-1-R and GRP78/BiP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed]
- Dekervel, J.; Bulle, A.; Windmolders, P.; Lambrechts, D.; Van Cutsem, E.; Verslype, C.; van Pelt, J. Acriflavine inhibits acquired drug resistance by blocking the epithelial-to-mesenchymal transition and the unfolded protein response. Transl. Oncol. 2017, 10, 59–69. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Factor | Material | Treatment (Concentration) | ER Stress Factors Involved | p | Brief | Ref |
|---|---|---|---|---|---|---|
| ATF6 | 45 patient samples (stage II and III) | none | ATF6α/P38 | =0.013 | ATF6α high expression and P38 low expression associates with poor outcome | [91] |
| GRP78 | 180 patient samples (stage I, II and III) | none | GRP78/CDK4/CDK6/STAT3/JAK2/RhoA | <0.05 | GRP78 overexpression associates with poor outcome | [57] |
| Cell lines (MIA PaCa-2, S2-VP10, SU.86.86) | Gemcitabine (400 nm), paclitaxel (50 nm), 5-FU (5 μm), GRP78 small interfering RNA (siRNA) | GRP78/ABC transporters | <0.001 | GRP78 downregulation diminishes chemoresistance and increases apoptosis when combined with chemotherapeutics | [59] | |
| Cell lines (Panc-1, CFPAC1, MIA-PaCa-2, Panc2.03, Panc02). Mouse samples (Panc-1 xenograft; Panc02 orthotopic implantation) | Cell lines: Erastin (2.5–40 μm), GRP78 short hairpin RNA (shRNA), ATF4 shRNA. Mice models: gemcitabine (20 mg/kg), sulfasalazine (SAS; 100 mg/kg/i.p.), gemcitabine + sulfasalazine, or gemcitabine + sulfasalazine + liproxstatin-1 (10 mg/kg/i.p) | ATF4/GRP78/GPX4 | 0.05 | GRP78 decreases ferroptosis and limits gemcitabine sensitivity both in vitro and in vivo | [103] | |
| Mice models (Pdx1-Cre; KrasG12D/+; p53f/+) | GRP78 genetically modified mice | GRP78 | <0.01 | GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia and tumorigenesis | [104] | |
| Cell lines (BxPC-3, Panc-1, Capan-1, Capan-2, CFPAC-1, HPAF-II, AsPC1). Mouse models (xenografts via subcutaneous injection) | ONC212 (cell lines: 20 μm; mice: 50 mg/kg) | GRP78/IGF1-R | <0.05 | ONC212 shows an anti-proliferative effect and induces apoptosis, reducing tumour growth by inducing UPR | [105] | |
| PERK | Cell lines (BxPC3). Mouse models (human tumour xenografts) | Cells lines: GSK2656157 (1 μmol/L), tunicamycin (5 μg/mL) and thapsigargin (1 μmol/L). Mice: GSK2656157 (50 or 150 mg/kg, orally) | PERK/EIF2α/ATF4/CHOP | <0.05 | PERK inhibition as a potential anti-tumour and anti-angiogenic approach | [65] |
| Cell lines (Panc-1, PK1, KLM1) | Avarol (40 μm) | GRP78/PERK/EIF2α/CHOP | <0.01 | Avarol induces apoptosis via CHOP | [55] | |
| P38 | 35 patient samples (stage I, II, III and IV). Cell lines (Panc5.04, Panc2.5, HPNE-E6/E7, HPDE). Mouse models (xenografts) | Cells lines: 5, 10, or 20 μmol/L. In vivo: intraperitoneal injection of SB202190 (2.5 mg/kg/day), and SP600125 (40 mg/kg/day) | P38/JNK | =0.041 | Active P38 contributes to better outcome | [96] |
| Cell lines (Panc-1), and derived cancer stem cells | Heparan sulfate hexasaccharide (100 μm) | P38/TCF4 | <0.005 | P38 activation inhibits cancer stem cells self-renewal inhibition | [97] | |
| ATF4 | Cell lines (AsPC-1, Panc-1) | ISRIB (250 nm), Gemcitabine (1 μm), ATF4 siRNA | EIF2/ATF4/CHOP | <0.01 | ATF4 downregulation induces apoptosis in combination of gemcitabine | [76] |
| Cell lines (Panc-1, HepG2, MIA PaCa-2) | Acriflavine (2.5 μm) | PERK/eIF2α/ATF4 | <0.001 | Acriflavine restores drug sensitivity by blocking UPR and EMT | [106] | |
| PERK/ATF6 | Mouse models (BxPC-3 xenografts) | Tanshinone IIA (0, 30 or 90 mg/kg) | PERK/ATF6/caspase-12/IRE1α/elF2α/p-JNK/CHOP/caspase-3 | <0.001 | Tan-IIA promotes apoptosis by induction of ER stress | [66] |
| Factor | Disease | Material | Treatment (Concentration) | ER Stress Factors Involved | p | Brief | Ref |
|---|---|---|---|---|---|---|---|
| ATF4 | Pancreatic neuroendocrine tumour | 45 patient samples | - | GRP78/ATF4/CHOP | <0.05 | ATF4 is overexpressed in pancreatic neuroendocrine tumours | [75] |
| MCL and AML | MCL cell lines (Z-138, JVM-2, MINO, and JeKo-1). AML cell lines (OCI-AML3, MOLM-13, HL-60, and THP-1). Primary cells. Mouse models (via tail vein injection) | ONC201 (5 μm), rapamycin (10 nm), or tunicamycin (1 μm) | ATF4/mTORC1 | <0.0001 | ONC201 induces apoptosis independent of TP53 mutation status and causes changes in gene expression similarly by UPR. ONC201 induces ATF4 and inhibits mTORC1 | [71] | |
| ATF4-ATF3-CHOP | TLL | TLL cell lines (Jurkat, Molt4) and the T-cell hybridoma cell line (DO11.10) | Farnesol (75 μm) | ATF4/ATF3/CHOP/PERK-eIF2α | <0.01 | Farnesol induces apoptosis in leukemic cells by induction of the PERK-eIF2α-ATF3/4 cascade | [72] |
| ATF4 | CRC | CRC cell lines (HCT116 and LoVo) | Glucose deprivation (1,5 mmol/L glucose) | GRP78/PERK/ATF4 | <0.001 | Glucose deprivation protects cells from oxaliplatin- and 5-fluorouracil-induced apoptosis, and induces the expression of ATF4. Depletion of ATF4 can induce apoptosis and drug re-sensitisation. | [73] |
| ATF6 | Insulinoma | Cell lines isolated from rat and mouse pancreas (INS-1 832/13) | Tunicamycin (0.1 µg/m), thapsigargin (0.1 µg/m), staurosporin, SB239063 (50 µm), and SP600125 (50 µm), ATF6α siRNA | GRP78/ATF6α | <0.05 | ATF6α knockdown activates JNK and P38 to induce apoptosis in insulinoma cells and primary islets | [90] |
| OC and CML | OC cell lines (HeLa), and CML cell lines (K562 and LAMA) | Dithiothreitol (1 mm), thapsigargin (500 nm), azetidine-2-carboxylic acid (10 mm), and tunicamycin (5 μg/mL) | PDIA5/ATF6/BiP | <0.01 | PDIA5/ATF6α axis modulates sensitivity of leukemia cells to imatinib | [92] | |
| IRE1α | AML | AML cell lines (NB4, U937, K-562, TF-1, HL-60, PL-21, and THP-1). Primary samples and murine hematopoietic cells | 2-hydroxy-1-naphthaldehyde (25 μm), STF-083010 (50 μm), and toyocamycin (500 nm) | IRE1α/XBP1 | <0.01 | Inhibition of IRE1α decreases cell viability and induces apoptosis and G1 cell cycle arrest | [81] |
| IRE1/ATF6 | Melanoma | Melanoma cell lines (Mel-RM, Mel-RMu, Mel-CV, and MM200) | siRNA and shRNA of IRE1α and ATF6 | IRE1α/ATF6 | <0.05 | IRE1α and ATF6 are critical for survival of melanoma cells undergoing ER stress | [93] |
| IRE1/XBP1 | BC | BC cell lines (MDA-MB-231 and MCF-7) | Thapsigargin (250 nm) or bortezomib (100 nm) | IRE1/XBP-1 | <0.05 | Estrogen receptor β sensitises BC cells to thapsigargin and to bortezomib by regulating the IRE1/XBP-1 pathway | [83] |
| BC | BC cell lines (SUM159, BT549, and MDA-MB-231), PDX models, and genetically engineered mouse models | Small molecule inhibitor 8866 (300 mg/kg oral daily) | IRE1/XBP1 pathway and MYC | <0.001 | Silencing of XBP1 selectively blocks the growth of MYC-hyperactivated cells. Pharmacological inhibition of IRE1 selectively restrained MYC-overexpressing tumour growth in vivo in a cohort of preclinical patient-derived xenograft models and genetically engineered mouse models | [84] | |
| XBP-1 | BC | CSC derived from MCF7 cell line (CD44+/CD24-) | Tunicamycin (2 μg/mL) | XBP-1/ATF6/CHOP | <0.001 | Tunicamycin inhibited invasion, increased cell death, suppressed proliferation, and reduced migration in a CD44+/CD24- and CD44+/CD24- rich MCF7 cell culture by an increased level of spliced XBP-1, ATF6 nuclear translocation and CHOP protein expression | [77] |
| P38α/β | CRC and BC | CRC cell lines (HT29, HCT116, and LS174T) and BC cell lines (MDA-MB-231) | Heparan sulfate hexasaccharide (100 µm) | TCF4 | <0.005 | Heparan sulfate hexasaccharide selectively inhibits CSC self-renewal and induces apoptosis in colorectal and breast CSCs | [97] |
| GRP78 | CRC | CRC cell lines (HT29, HT8, SW480 and colo205) | Oxaliplatin (5 µm) and vomitoxin (1 µg/mL) | GRP78/CD24 | <0.001 | Suppression of GRP78 sensitises human colorectal cancer cells to oxaliplatin by downregulation of CD24 | [62] |
| BC | BC cell lines (MCF-7 and T47D) | Plumbagin (from 0.5 to 5 μm) and Tamoxifen (1 or 5 μm) | GRP78/BIK | < 0.05 | Plumbagin inhibits GRP78 activity, and increases Bik expression and apoptosis induction, which contributes to the sensitisation of BC cells to tamoxifen | [63] | |
| NSCLC and GB | NSCLC cell lines (A549, and H460), and GB cell lines (D54 and U251). Mouse xenografts. | Anti-GRP78 antibody (1 μg/mL) | GRP78 and PI3K/AKT/mTOR signaling | <0.0001 | Anti-GRP78 attenuates cell proliferation, enhances radiation therapy, induces apoptosis, and delays tumour growth in mouse xenograft through the suppression of Akt/mTOR signaling | [64] | |
| PERK | Insulinoma | Mice samples (insulinoma generated by SV40 Large T-Antigen) | ISRIB (250 nm), Gemcitabine (1 μm), ATF4 siRNA | PERK | <0.000005 | PERK promotes tumour proliferation and angiogenesis | [68] |
| Breast, lung and gastric cancer | BC cell lines (MCF7, T47D, BT474, BT549, ZR-75–30, Hs578T, MDA-MB-157, and MDA-MB-231). Orthotopic injection into a mammary pad on NOD/SCID mice. Breast, lung and gastric cancer patient samples. | AEBSF (1 mg) | PERK/CREB3L1/ATF4 | < 0.01 | PERK signalling drives invasion and metastasis of breast cancer cell lines through CREB3L1, and associates with a poor outcome in breast, lung, and gastric cancer patients | [69] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Carbonero, N.; Li, W.; Cabeza-Morales, M.; Martinez-Useros, J.; Garcia-Foncillas, J. New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 2468. https://doi.org/10.3390/ijms19092468
Garcia-Carbonero N, Li W, Cabeza-Morales M, Martinez-Useros J, Garcia-Foncillas J. New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review. International Journal of Molecular Sciences. 2018; 19(9):2468. https://doi.org/10.3390/ijms19092468
Chicago/Turabian StyleGarcia-Carbonero, Nuria, Weiyao Li, Marticela Cabeza-Morales, Javier Martinez-Useros, and Jesus Garcia-Foncillas. 2018. "New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review" International Journal of Molecular Sciences 19, no. 9: 2468. https://doi.org/10.3390/ijms19092468