Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets




Abstract
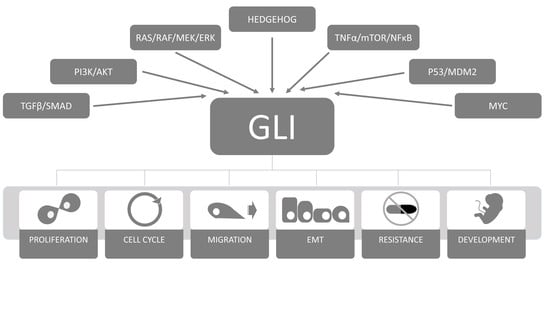
:
1. Introduction
2. The HH-GLI Signaling Pathway
2.1. Signaling at the Membrane
2.2. Cytoplasmic Signaling Cascade
2.3. Nuclear Signaling
2.4. The GLI Code
3. GLI Genes and Protein Isoforms
4. Role of GLI in Development
5. Role of GLI in Congenital Malformations and Syndromes
5.1. GLI1
5.2. GLI2
5.3. GLI3
6. Activation of GLI in Tumorigenesis
6.1. Ligand Dependent Signal Transduction
6.2. Ligand Independent Non-Canonical Activation and Cross-Talk with other Pathways
6.3. Genetic Changes
6.3.1. Gene Amplification
6.3.2. Gene Translocation (Fusion)
6.3.3. Short Genetic Variations
6.4. Epigenetic Changes
6.4.1. DNA/Protein/Histone Methylation
6.4.2. RNA Interference
7. GLI Proteins as Therapeutic Targets
7.1. GLI Inhibitors
7.2. Combining GLI Inhibitors with other Chemotherapeutic Agents
7.3. Role of GLI Proteins in Chemoresistance
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.; Outram, S.V.; Saldaña, J.I.; Furmanski, A.L.; Dessens, J.T.; Crompton, T. Regulation of murine normal and stress-induced erythropoiesis by Desert Hedgehog (Dhh). Blood 2012. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Varas, A.; Pepicelli, C.V.; Crompton, T. Hedgehog signaling regulates differentiation from double-negative to double-positive thymocyte. Immunity 2000, 13, 187–197. [Google Scholar] [CrossRef]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hager-Theodorides, A.L.; Furmanski, A.L.; Ross, S.E.; Outram, S.V.; Rowbotham, N.J.; Crompton, T. The Gli3 Transcription Factor Expressed in the Thymus Stroma Controls Thymocyte Negative Selection via Hedgehog-Dependent and -Independent Mechanisms. J. Immunol. 2009, 183, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu–Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [PubMed]
- Hui, C.; Angers, S. Gli Proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; Pasca di Magliano, M. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.T.; McMahon, A.P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999, 397, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.; Bijlsma, M.F.; Roelink, H. Shh-mediated degradation of Hhip allows cell autonomous and non-cell autonomous Shh signaling. Nat. Commun. 2014, 5, 4849. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Wang, W.; Hannoush, R.N.; de Sauvage, F.J. Regulation of the oncoprotein Smoothened by small molecules. Nat. Chem. Biol. 2015, 11, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.F.X.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.F.; Luchetti, G.; Rohatgi, R.; Siebold, C. Multiple ligand binding sites regulate the Hedgehog signal transducer Smoothened in vertebrates. Curr. Opin. Cell Biol. 2018, 51, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 2016, 5, e20304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Liu, Y.; Fan, J.; Zhang, J.; Li, X.-A.; Evers, B.M.; Zhu, H.; Jia, J. PI(4)P Promotes Phosphorylation and Conformational Change of Smoothened through Interaction with Its C-terminal Tail. PLoS Biol. 2016, 14, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 2013, 23, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yue, S.; Xie, L.; Pu, X.-H.; Jin, T.; Cheng, S.Y. Dual Phosphorylation of Suppressor of Fused (Sufu) by PKA and GSK3 Regulates Its Stability and Localization in the Primary Cilium. J. Biol. Chem. 2011, 286, 13502–13511. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Hwang, S.; Somatilaka, B.; Badgandi, H.; Jackson, P.K.; DeFea, K.; Mukhopadhyay, S. Smoothened determines β-arrestin–mediated removal of the G protein–coupled receptor Gpr161 from the primary cilium. J. Cell Biol. 2016, 212, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Gouti, M.; Sagner, A.; Sircar, R.; Luchetti, G.; Ingham, P.W.; Briscoe, J.; Rohatgi, R. G protein–coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci. Signal. 2018, 11, eaao5749. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.-Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog Signaling Is a Novel Therapeutic Target in Tamoxifen-Resistant Breast Cancer Aberrantly Activated by PI3K/AKT Pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, X.; Du, W.; Chen, L.; Wang, G.; Cui, Y.; Liu, Y.; Dou, Z.; Wang, H.; Zhang, P.; et al. Suppressor of fused (Sufu) represses Gli1 transcription and nuclear accumulation, inhibits glioma cell proliferation, invasion and vasculogenic mimicry, improving glioma chemo-sensitivity and prognosis. Oncotarget 2014, 5, 11681. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Han, Y.; Jiang, J. Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kapβ2 in Hedgehog signaling. J. Cell Sci. 2014, 127, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Tempe, D.; Casas, M.; Karaz, S.; Blanchet-Tournier, M.-F.; Concordet, J.-P. Multisite Protein kinase A and Glycogen synthase kinase 3 beta phosphorylation leads to Gli3 ubiquitination by SCF bTrCP. Mol. Cell. Biol. 2006, 26, 4316–4326. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Wang, Y.; Tang, J.; Luo, S. Molecular mechanisms of suppressor of fused in regulating the hedgehog signalling pathway. Oncol. Lett. 2018, 15, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5442–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paces-Fessy, M.; Boucher, D.; Petit, E.; Paute-Briand, S.; Blanchet-Tournier, M.-F. The negative regulator of Gli, Suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem. J. 2004, 378, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yao, E.; Wang, K.; Nozawa, Y.; Shimizu, H.; Johnson, J.R.; Chen, J.-N.; Krogan, N.J.; Chuang, P.-T. Regulation of Sufu activity by p66β and Mycbp provides new insight into vertebrate Hedgehog signaling. Genes Dev. 2014, 28, 2547–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Winklmayr, M.; Schmid, C.; Laner-Plamberger, S.; Kaser, A.; Aberger, F.; Eichberger, T.; Frischauf, A.M. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol. Biol. 2010, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. Context-dependent Regulation of the GLI Code in Cancer by HEDGEHOG and Non-HEDGEHOG Signals. J. Mol. Cell Biol. 2010, 2, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.-W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 13, 105. [Google Scholar] [PubMed]
- Speek, M.; Njunkova, O.; Pata, I.; Valdre, E.; Kogerman, P. A potential role of alternative splicing in the regulation of the transcriptional activity of human GLI 2 in gonadal tissues. BMC Mol. Biol. 2006, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L. From Normal Development to Disease: The Biochemistry and Regulation of GLI2. MEE 2014, 2, 1–19. [Google Scholar] [CrossRef]
- Démurger, F.; Ichkou, A.; Mougou-Zerelli, S.; Le Merrer, M.; Goudefroye, G.; Delezoide, A.-L.; Quélin, C.; Manouvrier, S.; Baujat, G.; Fradin, M.; et al. New insights into genotype-phenotype correlation for GLI3 mutations. Eur. J. Hum. Genet. 2015, 23, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Kalff-Suske, M.; Wild, A.; Topp, J.; Wessling, M.; Jacobsen, E.M.; Bornholdt, D.; Engel, H.; Heuer, H.; Aalfs, C.M.; Ausems, M.G.; et al. Point mutations throughout the GLI3 gene cause Greig cephalopolysyndactyly syndrome. Hum. Mol. Genet. 1999, 8, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Amable, L.; Gavin, E.; Kudo, K.; Meng, E.; Rocconi, R.P.; Shevde, L.A.; Reed, E. GLI1 upregulates C-JUN through a specific 130-kDa isoform. Int. J. Oncol. 2014, 44, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Tostar, U.; Lauth, M.; Palaniswamy, R.; Kasper, M.; Toftgård, R.; Zaphiropoulos, P.G. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. J. Biol. Chem. 2008, 283, 14345–14354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.-W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A Novel Splice Variant of GLI1 That Promotes Glioblastoma Cell Migration and Invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.-W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A. Gli proteins encode context-dependent positive and negative functions: Implications for development and disease. Development 1999, 126, 3205–3216. [Google Scholar] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roessler, E.; Ermilov, A.N.; Grange, D.K.; Wang, A.; Grachtchouk, M.; Dlugosz, A.A.; Muenke, M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 2005, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, P.W. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Matise, M.P.; Joyner, A.L. Gli genes in development and cancer. Oncogene 1999, 18, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Theil, T.; Kaesler, S.; Grotewold, L.; Böse, J.; Rüther, U. Gli genes and limb development. Cell Tissue Res. 1999, 296, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Drakopoulou, E.; Outram, S.V.; Rowbotham, N.J.; Ross, S.E.; Furmanski, A.L.; Saldana, J.I.; Hager-Theodorides, A.L.; Crompton, T. Non-redundant role for the transcription factor Gli1 at multiple stages of thymocyte development. Cell Cycle 2010, 9, 4144–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Furmanski, A.L.; Ross, S.E.; Outram, S.V.; Dessens, J.T.; Crompton, T. Sonic hedgehog negatively regulates pre-TCR–induced differentiation by a Gli2-dependent mechanism. Blood 2009, 113, 5144–5156. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Mo, R.; Shimamura, K.; Hui, C. Gli2 and Gli3 play distinct roles in the dorsoventral patterning of the mouse hindbrain. Dev. Biol. 2007, 302, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Bear, K.A.; Solomon, B.D.; Antonini, S.; Arnhold, I.J.P.; França, M.M.; Gerkes, E.H.; Grange, D.K.; Hadley, D.W.; Jääskeläinen, J.; Paulo, S.S.; et al. Pathogenic mutations in GLI2 cause a specific phenotype that is distinct from holoprosencephaly. J. Med. Genet. 2014, 51, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Hager-Theodorides, A.L.; Dessens, J.T.; Outram, S.V.; Crompton, T. The transcription factor Gli3 regulates differentiation of fetal CD4-CD8-Double Negative thymocytes. Blood 2005, 106, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Yanez, D.C.; Ross, S.; Lau, C.-I.; Papaioannou, E.; Li, J.; Saldaña, J.I.; Crompton, T. In the fetal thymus, Gli3 in thymic epithelial cells promotes thymocyte positive selection and differentiation by repression of Shh. Development 2018. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Lau, C.-I.; Saldaña, J.I.; Ross, S.; Crompton, T. The transcription factor Gli3 promotes B cell development in fetal liver through repression of Shh. J. Exp. Med. 2017, 214, 2041–2058. [Google Scholar] [CrossRef] [PubMed]
- Büscher, D.; Rüther, U. Expression profile of Gli family members and Shh in normal and mutant mouse limb development. Dev. Dyn. 1998, 211, 88–96. [Google Scholar] [CrossRef]
- Ahmed, H.; Akbari, H.; Emami, A.; Akbari, M.R. Genetic Overview of Syndactyly and Polydactyly. Plast. Reconstr. Surg. Glob. Open 2017, 5, e1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Li, Y.; Geng, J.; Fu, Q.; Xu, Y.; Shen, Y. Novel frame-shift mutations of GLI3 gene in non-syndromic postaxial polydactyly patients. Clin. Chim. Acta 2014, 433, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Campos, A.; Ullah, A.; Nevado, J.; Yildirim, R.; Unal, E.; Ciorraga, M.; Barruz, P.; Chico, L.; Piceci-Sparascio, F.; Guida, V.; et al. GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum. Mol. Genet. 2017, 26, 4556–4571. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M. Holoprosencephaly: Clinical, anatomic, and molecular dimensions. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Bertolacini, C.D.P.; Ribeiro-Bicudo, L.A.; Petrin, A.; Richieri-Costa, A.; Murray, J.C. Clinical findings in patients with GLI2 mutations—Phenotypic variability. Clin. Genet. 2012, 81, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Du, Y.-Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer Hovinga, I.C.L.; Giltay, J.C.; van der Crabben, S.N.; Steyls, A.; van der Kamp, H.J.; Paulussen, A.D.C. Extreme phenotypic variability of a novel GLI2 mutation in a large family with panhypopituitarism and polydactyly: Clinical implications. Clin. Endocrinol. (Oxf.) 2018. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Ribeiro, L.A.; de Miranda, E.; Richieri-Costa, A.; Murray, J.C. GLI2 mutations in four Brazilian patients: How wide is the phenotypic spectrum? Am. J. Med. Genet. Part A 2006, 140A, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, P.; Schoumans, J.; Staaf, J.; Jönsson, G.; Carlsson, F.; Kristoffersson, U.; Borg, A.; Nordenskjöld, M.; Dahl, N. Hemizygosity for chromosome 2q14.2-q22.1 spanning the GLI2 and PROC genes associated with growth hormone deficiency, polydactyly, deep vein thrombosis and urogenital abnormalities. Clin. Genet. 2006, 69, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Kevelam, S.H.G.; van Harssel, J.J.T.; van der Zwaag, B.; Smeets, H.J.M.; Paulussen, A.D.C.; Lichtenbelt, K.D. A patient with a mild holoprosencephaly spectrum phenotype and heterotaxy and a 1.3 Mb deletion encompassing GLI2. Am. J. Med. Genet. Part A 2011, 158A, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Greally, M.T.; Robinson, E.; Allen, N.M.; O’Donovan, D.; Crolla, J.A. De novo interstitial deletion 2q14.1q22.1: Is there a recognizable phenotype? Am. J. Med. Genet. A 2014, 164A, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Marion, R.; Punjabi, N.P.; Pereira, E.; Samanich, J.; Agarwal, C.; Li, J.; Huang, C.-K.; Ramesh, K.H.; Cannizzaro, L.A.; et al. A de novo 10.79 Mb interstitial deletion at 2q13q14.2 involving PAX8 causing hypothyroidism and mullerian agenesis: A novel case report and literature review. Mol. Cytogenet. 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Kordaß, U.; Schröder, C.; Elbracht, M.; Soellner, L.; Eggermann, T. A familial GLI2 deletion (2q14.2) not associated with the holoprosencephaly syndrome phenotype. Am. J. Med. Genet. A 2015, 167A, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Goumy, C.; Gay-Bellile, M.; Salaun, G.; Kemeny, S.; Eymard-Pierre, E.; Biard, M.; Pebrel-Richard, C.; Vanlieferinghen, P.; Francannet, C.; Tchirkov, A.; et al. A novel 2q14.1q14.3 deletion involving GLI2 and RNU4ATAC genes associated with partial corpus callosum agenesis and severe intrauterine growth retardation. Birth Defects Res. Part A Clin. Mol. Teratol. 2016, 106, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Niida, Y.; Inoue, M.; Ozaki, M.; Takase, E. Human Malformation Syndromes of Defective GLI: Opposite Phenotypes of 2q14.2 (GLI2) and 7p14.2 (GLI3) Microdeletions and a GLIA/R Balance Model. Cytogenet. Genome Res. 2017, 153, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. The Greig cephalopolysyndactyly syndrome. Orphanet J. Rare Dis. 2008, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Volodarsky, M.; Langer, Y.; Birk, O.S. A novel GLI3 mutation affecting the zinc finger domain leads to preaxial-postaxial polydactyly-syndactyly complex. BMC Med. Genet. 2014, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Speksnijder, L.; Cohen-Overbeek, T.E.; Knapen, M.F.C.M.; Lunshof, S.M.; Hoogeboom, A.J.M.; van den Ouwenland, A.M.; de Coo, I.F.M.; Lequin, M.H.; Bolz, H.J.; Bergmann, C.; et al. A de novo GLI3 mutation in a patient with acrocallosal syndrome. Am. J. Med. Genet. A 2013, 161A, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Al-Qattan, M.M.; Shamseldin, H.E.; Salih, M.A.; Alkuraya, F.S. GLI3-related polydactyly: A review. Clin. Genet. 2017, 92, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Pallister-Hall Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Huang, S.Y.; Yang, J.-Y. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers (Basel) 2015, 7, 2110–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecher, L.A.; Sharfman, W.H. Advanced basal cell carcinoma, the hedgehog pathway, and treatment options—Role of smoothened inhibitors. Biologics 2015, 9, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, D.; Jiang, J.; Hu, J.; Zhang, W.; Chen, Y.; Cui, X.; Qi, Y.; Zou, H.; Zhang, W.; et al. Analysis of molecular cytogenetic alteration in rhabdomyosarcoma by array comparative genomic hybridization. PLoS ONE 2014, 9, e94924. [Google Scholar] [CrossRef] [PubMed]
- Duman-Scheel, M.; Weng, L.; Xin, S.; Du, W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 2002, 417, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Bigelow, R.L.H. Transcriptional Regulation of bcl-2 Mediated by the Sonic Hedgehog Signaling Pathway through gli-1. J. Biol. Chem. 2003, 279, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pola, R.; Ling, L.E.; Silver, M.; Corbley, M.J.; Kearney, M.; Blake Pepinsky, R.; Shapiro, R.; Taylor, F.R.; Baker, D.P.; Asahara, T.; et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med. 2001, 7, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Talbot, L.J.; Bhattacharya, S.D.; Kuo, P.C. Epithelial-mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. Int. J. Biochem. Mol. Biol. 2012, 3, 117–136. [Google Scholar] [PubMed]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non–Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S. Nanog, Gli, and p53: A new network of stemness in development and cancer. EMBO J. 2010, 29, 2475–2476. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Deb, M.; Sengupta, D.; Shilpi, A.; Bhutia, S.K.; Patra, S.K. Intricacies of hedgehog signaling pathways: A perspective in tumorigenesis. Exp. Cell Res. 2012, 318, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta (BBA) Rev. Cancer 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Goetz, J.A.; Singh, S.; Ogden, S.K.; Petty, W.J.; Black, C.C.; Memoli, V.A.; Dmitrovsky, E.; Robbins, D.J. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene 2006, 26, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Qualtrough, D.; Buda, A.; Gaffield, W.; Williams, A.C.; Paraskeva, C. Hedgehog signalling in colorectal tumour cells: Induction of apoptosis with cyclopamine treatment. Int. J. Cancer 2004, 110, 831–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernández, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differs in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.G.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. HEDGEHOG-GLI Signaling Drives Self-Renewal and Tumorigenicity of Human Melanoma-Initiating Cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational Prediction and Experimental Verification of New MAP Kinase Docking Sites and Substrates Including Gli Transcription Factors. PLoS Comput. Biol. 2010, 6, e1000908. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS Activates Hedgehog Signaling Pathway in Pancreatic Cancer Cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Li, J.; Gao, T.; Xie, J.; Evers, B.M. Protein Kinase Cδ Negatively Regulates Hedgehog Signaling by Inhibition of Gli1 Activity. J. Biol. Chem. 2009, 284, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated b cells (NF-κB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Montagnani, V.; Penachioni, J.Y.; Vinci, M.C.; Olivito, B.; Borgognoni, L.; Stecca, B. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 2013, 32, 4737–4747. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers (Basel) 2015, 7, 1684–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.G.; Iannaccone, S.; Vieux, K.-F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Flora, A.; Klisch, T.J.; Schuster, G.; Zoghbi, H.Y. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science 2009, 326, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta 2015, 1852, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Rahman, M.F.-U.; Tostar, U.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzari, E.; Mondala, P.K.; Santos, N.D.; Miller, A.C.; Pineda, G.; Jiang, Q.; Leu, H.; Ali, S.A.; Ganesan, A.-P.; Wu, C.N.; et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 2017, 8, 1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.; Bigner, S.; Bigner, D.; Trent, J.; Law, M.; O’Brien, S.; Wong, A.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, T.L.; Greshock, J.; Wang, Y.; Colligon, T.; Yu, Q.C.; Clemmer, V.; Zaks, T.Z.; Weber, B.L. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005, 7, R1186–R1198. [Google Scholar] [CrossRef] [PubMed]
- Nessling, M.; Richter, K.; Schwaenen, C.; Roerig, P.; Wrobel, G.; Wessendorf, S.; Fritz, B.; Bentz, M.; Sinn, H.-P.; Radlwimmer, B.; et al. Candidate genes in breast cancer revealed by microarray-based comparative genomic hybridization of archived tissue. Cancer Res. 2005, 65, 439–447. [Google Scholar] [PubMed]
- Roberts, W.M.; Douglass, E.C.; Peiper, S.C.; Houghton, P.J.; Look, A.T. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989, 49, 5407–5413. [Google Scholar] [PubMed]
- Snijders, A.M.; Schmidt, B.L.; Fridlyand, J.; Dekker, N.; Pinkel, D.; Jordan, R.C.K.; Albertson, D.G. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 2005, 24, 4232–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlén, A.; Mertens, F.; Mandahl, N.; Panagopoulos, I. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12). Biochem. Biophys. Res. Commun. 2004, 325, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A.; Sanders, K.; Huang, D.; Nelson, M.; Neff, J.R.; Muirhead, D.; Walker, C.; Seemayer, T.A.; Sumegi, J. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum. Pathol. 2012, 43, 1524–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuromi, T.; Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Akizuki, G.; Kitamura, Y.; Hayashi, K. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum. Pathol. 2017, 69, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, e102–e105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Song, S.Y.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Intratumoral Heterogeneity of Frameshift Mutations of GLI1 Encoding a Hedgehog Signaling Protein in Colorectal Cancers. Pathol. Oncol. Res. 2018, 24, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, C.; Khalifa, E.; Laizet, Y.; Soubeyran, I.; Mathoulin-Pelissier, S.; Chomienne, C.; Italiano, A. Targetable Alterations in Adult Patients With Soft-Tissue Sarcomas: Insights for Personalized Therapy. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zuo, Y. Occurrence of HHIP gene CpG island methylation in gastric cancer. Oncol. Lett. 2014, 8, 2340–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretnik, M.; Musani, V.; Oreskovic, S.; Leovic, D.; Levanat, S. The Patched gene is epigenetically regulated in ovarian dermoids and fibromas, but not in basocellular carcinomas. Int. J. Mol. Med. 2007, 19, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löf-Öhlin, Z.M.; Levanat, S.; Sabol, M.; Sorbe, B.; Nilsson, T.K. Promoter methylation in the PTCH gene in cervical epithelial cancer and ovarian cancer tissue as studied by eight novel Pyrosequencing® assays. Int. J. Oncol. 2011, 38, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Quan, H.; Xie, C.; Lou, L. NL-103, a novel dual-targeted inhibitor of histone deacetylases and hedgehog pathway, effectively overcomes vismodegib resistance conferred by Smo mutations. Pharmacol. Res. Perspect. 2014, 2, e00043. [Google Scholar] [CrossRef] [PubMed]
- Levanat, S.; Sabol, M.; Musani, V.; Ozretic, P.; Trnski, D. Hedgehog signaling pathway as genetic and epigenetic target in ovarian tumors. Curr. Pharm. Des. 2017, 23, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.; Liem, N.; Vaithilingam, A.; Lim, P.L.; Sapari, N.S.; Elahi, E.; Mok, Z.Y.; Cheng, C.L.; Yan, B.; Pang, B.; et al. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: A comprehensive profiling approach. BMC Gastroenterol. 2014, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Lorente, A.; Castresana, J.S. Hedgehog signalling in medulloblastoma, glioblastoma and neuroblastoma. Oncol. Rep. 2008, 19, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, J.; Tian, T.; Fu, X.; Wang, W.; Li, S.; Shi, T.; Suo, A.; Ruan, Z.; Guo, H.; et al. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumor Biol. 2016, 37, 9311–9322. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wu, H.; Cheng, S.Y.; Gao, D.; Zhang, L.; Zhao, Y. Set7 mediated Gli3 methylation plays a positive role in the activation of Sonic Hedgehog pathway in mammals. Elife 2016, 5, e15690. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Rahman, M.F.-U.; Fernandez-Barrena, M.G.; Diao, Y.; Liapi, E.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Annaratone, L.; Sapino, A.; et al. Identification of novel non-coding RNA-based negative feedback regulating the expression of the oncogenic transcription factor GLI1. Mol. Oncol. 2014, 8, 912–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, E.; De Smaele, E.; Miele, E.; Laneve, P.; Po, A.; Pelloni, M.; Paganelli, A.; Di Marcotullio, L.; Caffarelli, E.; Screpanti, I.; et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 2008, 27, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, E.; Smaele, E.D.; Po, A.; Marcotullio, L.D.; Tosi, E.; Espinola, M.S.B.; Rocco, C.D.; Riccardi, R.; Giangaspero, F.; Farcomeni, A.; et al. MicroRNA profiling in human medulloblastoma. Int. J. Cancer 2009, 124, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, S.; Birks, D.K.; Balakrishnan, I.; Alimova, I.; Harris, P.S.; Patel, P.R.; Handler, M.H.; Dubuc, A.; Taylor, M.D.; Foreman, N.K.; et al. MicroRNA 218 Acts as a Tumor Suppressor by Targeting Multiple Cancer Phenotype-associated Genes in Medulloblastoma. J. Biol. Chem. 2013, 288, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Li, D.; Qin, M.; Luo, D.; Zhang, X.; Zhao, H.; Hu, S. MicroRNA218 inhibits glioma migration and invasion via inhibiting glioma-associated oncogene homolog 1 expression at N terminus. Tumor Biol. 2014, 35, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, C.; Wang, M.; Su, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; et al. Decrease of miR-202-3p Expression, a Novel Tumor Suppressor, in Gastric Cancer. PLoS ONE 2013, 8, e69756. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, J.; Zhang, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; Zhu, Z. MiR-133b is frequently decreased in gastric cancer and its overexpression reduces the metastatic potential of gastric cancer cells. BMC Cancer 2014, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Sand, M.; Skrygan, M.; Sand, D.; Georgas, D.; Hahn, S.A.; Gambichler, T.; Altmeyer, P.; Bechara, F.G. Expression of microRNAs in basal cell carcinoma. Br. J. Dermatol. 2012, 167, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Fang, Z.; Cui, S.; Zhang, X.; Wu, Y.; Tang, H.; Tu, Y.; Ding, Y. Thermo-chemotherapy Induced miR-218 upregulation inhibits the invasion of gastric cancer via targeting Gli2 and E-cadherin. Tumor Biol. 2015, 36, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Constantin, L.; Wainwright, B.J. MicroRNAs Promote Granule Cell Expansion in the Cerebellum Through Gli2. Cerebellum 2015, 14, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Li, K.K.-W.; Xia, T.; Ma, F.M.T.; Zhang, R.; Mao, Y.; Wang, Y.; Zhou, L.; Lau, K.-M.; Ng, H.-K. miR-106b is overexpressed in medulloblastomas and interacts directly with PTEN. Neuropathol. Appl. Neurobiol. 2015, 41, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Cushing, L.; Ai, X.; Lü, J. miR-326 Is Downstream of Sonic Hedgehog Signaling and Regulates the Expression of Gli2 and Smoothened. Am. J. Respir. Cell Mol. Biol. 2014, 51, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, F.; Zheng, Y.; Zheng, Y.; Hong, W.; Hong, W.; Chen, B.; Chen, B.; Dong, P.; Dong, P.; et al. MicroRNA-200a suppresses epithelial-to-mesenchymal transition in rat hepatic stellate cells via GLI family zinc finger 2. Mol. Med. Rep. 2015, 12, 8121–8128. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wang, J.; Chen, M.; Chen, G.; Wu, Z.; Ying, L.; Zhuo, Q.; Zhang, J.; Wang, W. miR-200a suppresses cell growth and migration by targeting MACC1 and predicts prognosis in hepatocellular carcinoma. Oncol. Rep. 2015, 33, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.-S.; Kim, S.-W.; Yun, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 10993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, Z.; Li, M.-J.; Guo, H.-Y.; Jing, N.-C. Long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 regulates the expression of Gli2 by miR-202 to strengthen gastric cancer progression. Biomed. Pharmacother. 2017, 85, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Lee, S.T.; Welch, K.D.; Panter, K.E.; Gardner, D.R.; Garrossian, M.; Chang, C.-W.T. Cyclopamine: From Cyclops Lambs to Cancer Treatment. J. Agric. Food Chem. 2014, 62, 7355–7362. [Google Scholar] [CrossRef] [PubMed]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Ferretti, E.; Gulino, A. Vismodegib, a small-molecule inhibitor of the hedgehog pathway for the treatment of advanced cancers. Curr. Opin. Investig. Drugs 2010, 11, 707–718. [Google Scholar] [PubMed]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II Evaluation of IPI-926, an Oral Hedgehog Inhibitor, in Patients with Myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.C.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevåg, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.B.; Prêle, C.M.; Baltic, S.; Arthur, P.G.; Creaney, J.; Watkins, D.N.; Thompson, P.J.; Mutsaers, S.E. Mitochondria-derived reactive oxygen species drive GANT61-induced mesothelioma cell apoptosis. Oncotarget 2015, 6, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Xu, Y.; An, Y.; An, Y.; Wang, X.; Wang, X.; Zha, W.; Zha, W.; Li, X.; Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 2014, 31, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog Signaling Regulates Telomerase Reverse Transcriptase in Human Cancer Cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnhold, V.; Boos, J.; Lanvers-Kaminsky, C. Targeting hedgehog signaling pathway in pediatric tumors: In vitro evaluation of SMO and GLI inhibitors. Cancer Chemother. Pharmacol. 2016, 77, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Moshai, E.F.; Wémeau-Stervinou, L.; Cigna, N.; Brayer, S.; Sommé, J.M.; Crestani, B.; Mailleux, A.A. Targeting the Hedgehog–Glioma-Associated Oncogene Homolog Pathway Inhibits Bleomycin-Induced Lung Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. HEDGEHOG-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Toftgård, R.; Zaphiropoulos, P.G.; Shimokawa, T. Reduction of Human Embryonal Rhabdomyosarcoma Tumor Growth by Inhibition of the Hedgehog Signaling Pathway. Genes Cancer 2010, 1, 941–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesslich, T.; Mayr, C.; Wachter, J.; Bach, D.; Fuereder, J.; Wagner, A.; Alinger, B.; Pichler, M.; Fazio, P.D.; Ocker, M.; et al. Activated hedgehog pathway is a potential target for pharmacological intervention in biliary tract cancer. Mol. Cell. Biochem. 2014, 396, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Desch, P.; Asslaber, D.; Kern, D.; Schnidar, H.; Mangelberger, D.; Alinger, B.; Stoecher, M.; Hofbauer, S.W.; Neureiter, D.; Tinhofer, I.; et al. Inhibition of GLI, but not Smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene 2010, 29, 4885. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Qiu, L.; Qi, M.; Liu, J.; Hu, K.; Lin, W.; Huang, Y.; Fu, J. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. J. Cell. Biochem. 2018, 119, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Wang, L.-X.; Zheng, F.-M.; Lai, S.-P.; Xu, D.-R.; Hu, Y.; Lin, D.-J.; Zhang, X.-Z.; Dong, L.; Long, Z.-J.; et al. Targeting GLI1 Suppresses Cell Growth and Enhances Chemosensitivity in CD34+ Enriched Acute Myeloid Leukemia Progenitor Cells. CPB 2016, 38, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Iwasawa, S.; Ebata, T.; Fan, M.; Tada, Y.; Tatsumi, K.; Takiguchi, Y. Inhibition of Gli leads to antitumor growth and enhancement of cisplatin-induced cytotoxicity in large cell neuroendocrine carcinoma of the lung. Oncol. Rep. 2018, 39, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hu, L.; Liu, Z.; Qin, Y.; Li, R.; Zhang, G.; Zhao, B.; Bi, C.; Lei, Y.; Bai, Y. Inhibition of Gli1-mediated prostate cancer cell proliferation by inhibiting the mTOR/S6K1 signaling pathway. Oncol. Lett. 2017, 14, 7970–7976. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, J.; Latuske, E.; Köhler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gu, S.; Huang, J.; Chen, S.; Zhang, Z.; Xu, M. Inhibition of autophagy potentiates the efficacy of Gli inhibitor GANT-61 in MYCN-amplified neuroblastoma cells. BMC Cancer 2014, 14, 768. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Tabata, K.; Suzuki, T. The GANT61, a GLI inhibitor, induces caspase-independent apoptosis of SK-N-LO cells. Biol. Pharm. Bull. 2014, 37, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, R.; Zeng, C.; Lu, Q.; Huang, D.; Shi, C.; Zhang, W.; Deng, L.; Yan, R.; Rao, H.; et al. Down-regulation of Gli transcription factor leads to the inhibition of migration and invasion of ovarian cancer cells via integrin β4-mediated FAK signaling. PLoS ONE 2014, 9, e88386. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Lu, K.; Li, P.; Li, X.; Zhou, X.; Li, Y.; Wang, X. GLI1 inhibitor GANT61 exhibits antitumor efficacy in T-cell lymphoma cells through down-regulation of p-STAT3 and SOCS3. Oncotarget 2016, 8, 48701–48710. [Google Scholar] [CrossRef] [PubMed]
- Samarzija, I.; Beard, P. Hedgehog pathway regulators influence cervical cancer cell proliferation, survival and migration. Biochem. Biophysical Res. Commun. 2012, 425, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Réda, J.; Ondrušová, L.; Krayem, M.; Ghanem, G.; Vachtenheim, J. GLI inhibitor GANT61 kills melanoma cells and acts in synergy with obatoclax. Int. J. Oncol. 2016, 49, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sang, F.; Chang, J.; Hua, Y.; Shi, W.; Tang, L.; Liu, L. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. Onco Targets Ther. 2013, 6, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Nagano, S.; Nagao, H.; Ishidou, Y.; Yokouchi, M.; Abematsu, M.; Yamamoto, T.; Komiya, S.; Setoguchi, T. Arsenic Trioxide Prevents Osteosarcoma Growth by Inhibition of GLI Transcription via DNA Damage Accumulation. PLoS ONE 2013, 8, e69466. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Setoguchi, T.; Nagata, M.; Tsuru, A.; Nakamura, S.; Nagano, S.; Ishidou, Y.; Nagao-Kitamoto, H.; Yokouchi, M.; Maeda, S.; et al. Combination of Hedgehog inhibitors and standard anticancer agents synergistically prevent osteosarcoma growth. Int. J. Oncol. 2016, 48, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.J.; Guo, Y.J.; Gao, Y.R.; Li, S.; Dai, Z.H.; Dong, X.Q.; Xu, Y.F.; Liu, C.Q.; Liu, Z.Y. Synergism between arsenic trioxide and cyclopamine in the inhibition of PC3 cell survival via the Hedgehog signaling pathway. Neoplasma 2015, 62, 894–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerl, K.; Moreno, N.; Holsten, T.; Ahlfeld, J.; Mertins, J.; Hotfilder, M.; Kool, M.; Bartelheim, K.; Schleicher, S.; Handgretinger, R.; et al. Arsenic trioxide inhibits tumor cell growth in malignant rhabdoid tumors in vitro and in vivo by targeting overexpressed Gli1. Int. J. Cancer 2014, 135, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Zhou, L.-F.; Gao, L.-J.; Wei, Y.; Xu, S.-F.; Chen, F.-Y.; Huang, W.-J.; Tan, W.-F.; Ye, Y.-P. Cynanbungeigenin C and D, a pair of novel epimers from Cynanchum bungei, suppress hedgehog pathway-dependent medulloblastoma by blocking signaling at the level of Gli. Cancer Lett. 2018, 420, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Subramani, R.; Gonzalez, E.; Nandy, S.B.; Arumugam, A.; Camacho, F.; Medel, J.; Alabi, D.; Lakshmanaswamy, R. Gedunin inhibits pancreatic cancer by altering sonic hedgehog signaling pathway. Oncotarget 2016, 8, 10891–10904. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lui, N.; Cheng, T.; Tseng, H.-H.K.; Yue, D.; Giroux-Leprieur, E.; Do, H.T.; Sheng, Q.; Jin, J.Q.; Luh, T.W.; et al. Gli as a Novel Therapeutic Target in Malignant Pleural Mesothelioma. PLoS ONE 2013, 8, e57346. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.A.; Uchida, K.; Sadhu, S.K.; Ahmed, F.; Ishibashi, M. Physalin H from Solanum nigrum as an Hh signaling inhibitor blocks GLI1–DNA-complex formation. Beilstein J. Org. Chem. 2014, 10, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, M.A.; Fujimatsu, T.; Uchida, K.; Sadhu, S.K.; Ahmed, F.; Ishibashi, M. Hh signaling inhibitors from Vitex negundo; naturally occurring inhibitors of the GLI1-DNA complex. Mol. Biosyst. 2013, 9, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, W.; Tan, W. Solasonine, A Natural Glycoalkaloid Compound, Inhibits Gli-Mediated Transcriptional Activity. Molecules 2016, 21, 1364. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, K.-S.; Jeng, C.-J.; Sheen, I.-S.; Wu, S.-H.; Lu, S.-J.; Wang, C.-H.; Chang, C.-F. Glioma-Associated Oncogene Homolog Inhibitors Have the Potential of Suppressing Cancer Stem Cells of Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1375. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Sun, B.; Li, Z.; Lin, L.; Meng, X.; Han, B.; Wang, R.; Wu, P.; Li, J.; Cai, J.; et al. Aspirin inhibits the SHH/GLI1 signaling pathway and sensitizes malignant glioma cells to temozolomide therapy. Aging (Albany NY) 2017, 9, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Shin, H.-S.; Lee, Y.S.; Lee, D.; Kim, S.; Lee, Y.C. Genistein attenuates cancer stem cell characteristics in gastric cancer through the downregulation of Gli1. Oncol. Rep. 2014, 31, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein decreases the breast cancer stem-like cell population through Hedgehog pathway. Stem Cell Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, T.N.; McLaughlin, E.A.; Abdel-Hamid, M.K.; Gordon, C.P.; Bernstein, I.R.; Pye, V.; Cossar, P.; Sakoff, J.A.; McCluskey, A. Quinolone-1-(2H)-ones as hedgehog signalling pathway inhibitors. Org. Biomol. Chem. 2016, 14, 6304–6315. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Huang, W.; Wang, J.; Liu, X.; Yang, J.; Zhang, Y.; Geng, Y.; Tan, W.; Zhang, A. Discovery of Novel Macrocyclic Hedgehog Pathway Inhibitors Acting by Suppressing the Gli-Mediated Transcription. J. Med. Chem. 2017, 60, 8218–8245. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, H.; Oue, T.; Uehara, S.; Fukuzawa, M. Forskolin, a Hedgehog signal inhibitor, inhibits cell proliferation and induces apoptosis in pediatric tumor cell lines. Mol. Med. Rep. 2010, 3, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Shen, X.; Cheng, L.; Hong, D.; Chen, B. Perifosine inhibits S6K1-Gli1 signaling and enhances gemcitabine-induced anti-pancreatic cancer efficiency. Cancer Chemother. Pharmacol. 2014, 73, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Siddharth, S.; Das, S.; Nayak, D.; Sethy, C.; Kundu, C.N. Nanoquinacrine caused apoptosis in oral cancer stem cells by disrupting the interaction between GLI1 and β catenin through activation of GSK3β. Toxicol. Appl. Pharmacol. 2017, 330, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic Trioxide Inhibits the Hedgehog Pathway Which Is Aberrantly Activated in Acute Promyelocytic Leukemia. AHA 2013, 130, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanyamraju, P.K.; Holz, P.S.; Finkernagel, F.; Fendrich, V.; Lauth, M. Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway. Mol. Cancer Ther. 2015, 14, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Ingallina, C.; Costa, P.M.; Ghirga, F.; Klippstein, R.; Wang, J.T.; Berardozzi, S.; Hodgins, N.; Infante, P.; Pollard, S.M.; Botta, B.; et al. Polymeric glabrescione B nanocapsules for passive targeting of Hedgehog-dependent tumor therapy in vitro. Nanomedicine (London) 2017, 12, 711–728. [Google Scholar] [CrossRef] [PubMed]
- Chenna, V.; Hu, C.; Pramanik, D.; Aftab, B.T.; Karikari, C.; Campbell, N.R.; Hong, S.-M.; Zhao, M.; Rudek, M.A.; Khan, S.R.; et al. A Polymeric Nanoparticle Encapsulated Small Molecule Inhibitor of Hedgehog Signaling (NanoHHI) Bypasses Secondary Mutational Resistance to Smoothened Antagonists. Mol. Cancer Ther. 2012, 11, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Sabol, M.; Trnski, D.; Uzarevic, Z.; Ozretic, P.; Musani, V.; Rafaj, M.; Cindric, M.; Levanat, S. Combination of Cyclopamine and Tamoxifen Promotes Survival and Migration of MCF-7 Breast Cancer Cells—Interaction of Hedgehog-Gli and Estrogen Receptor Signaling Pathways. PLoS ONE 2014, 9, e114510. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Azatyan, A.; Rahman, M.F.-U.; Zhao, C.; Zhu, J.; Dahlman-Wright, K.; Zaphiropoulos, P.G. Blockade of the Hedgehog pathway downregulates estrogen receptor alpha signaling in breast cancer cells. Oncotarget 2016, 7, 71580–71593. [Google Scholar] [CrossRef] [PubMed]
- Paret, C.; Russo, A.; Otto, H.; Mayer, A.; Zahnreich, S.; Wagner, W.; Samuel, D.; Scharnhorst, D.; Solomon, D.A.; Dhall, G.; et al. Personalized therapy: CNS HGNET-BCOR responsiveness to arsenic trioxide combined with radiotherapy. Oncotarget 2017, 8, 114210–114225. [Google Scholar] [CrossRef] [PubMed]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Latuske, E.-M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jücker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurebayashi, J.; Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017, 108, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Muschel, R.J.; Haustermans, K. The Effect of Metformin and GANT61 Combinations on the Radiosensitivity of Prostate Cancer Cells. Int. J. Mol. Sci. 2017, 18, 399. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Farheen, S.; Mariyath, M.P.M.; Castresana, J.S. Potential role of Shh-Gli1-BMI1 signaling pathway nexus in glioma chemoresistance. Tumour Biol. 2016, 37, 15107–15114. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, P.; Satapathy, S.R.; Siddharth, S.; Das, D.; Nayak, A.; Kundu, C.N. Resveratrol and curcumin synergistically induces apoptosis in cigarette smoke condensate transformed breast epithelial cells through a p21(Waf1/Cip1) mediated inhibition of Hh-Gli signaling. Int. J. Biochem. Cell Biol. 2015, 66, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets Ther. 2015, 8, 877–883. [Google Scholar] [CrossRef]
- Pan, D.; Li, Y.; Li, Z.; Wang, Y.; Wang, P.; Liang, Y. Gli inhibitor GANT61 causes apoptosis in myeloid leukemia cells and acts in synergy with rapamycin. Leuk. Res. 2012, 36, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Graab, U.; Hahn, H.; Fulda, S. Identification of a novel synthetic lethality of combined inhibition of hedgehog and PI3K signaling in rhabdomyosarcoma. Oncotarget 2015, 6, 8722–8735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebenko, M.; Drenckhan, A.; Gros, S.J.; Jücker, M.; Grabinski, N.; Ewald, F.; Grottke, A.; Schultze, A.; Izbicki, J.R.; Bokemeyer, C.; et al. ErbB2 signaling activates the Hedgehog pathway via PI3K-Akt in human esophageal adenocarcinoma: Identification of novel targets for concerted therapy concepts. Cell. Signal. 2015, 27, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Shishido, S.M.; Queiroz, K.C.S.; Oliveira, D.N.; Faria, A.L.C.; Catharino, R.R.; Spek, C.A.; Ferreira, C.V. Irradiated Riboflavin Diminishes the Aggressiveness of Melanoma In Vitro and In Vivo. PLoS ONE 2013, 8, e54269. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Rom, W.N.; Koshiji, M.; Mo, Y.; Hosomi, Y.; Tchou-Wong, K.-M. Role of GLI1 and NDRG1 in Increased Resistance to Apoptosis Induction. J. Environ. Pathol. Toxicol. Oncol. 2015, 34, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, W.; Li, J.; Zhang, J.; Wang, X.; Wang, M. Sulforaphane reverses gefitinib tolerance in human lung cancer cells via modulation of sonic hedgehog signaling. Oncol. Lett. 2018, 15, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Armas-López, L.; Piña-Sánchez, P.; Arrieta, O.; de Alba, E.G.; Ortiz-Quintero, B.; Santillán-Doherty, P.; Christiani, D.C.; Zúñiga, J.; Ávila-Moreno, F. Epigenomic study identifies a novel mesenchyme homeobox2-GLI1 transcription axis involved in cancer drug resistance, overall survival and therapy prognosis in lung cancer patients. Oncotarget 2017, 8, 67056–67081. [Google Scholar] [CrossRef] [PubMed]
- Giroux Leprieur, E.; Vieira, T.; Antoine, M.; Rozensztajn, N.; Rabbe, N.; Ruppert, A.-M.; Lavole, A.; Cadranel, J.; Wislez, M. Sonic Hedgehog Pathway Activation Is Associated with Resistance to Platinum-Based Chemotherapy in Advanced Non-Small-Cell Lung Carcinoma. Clin. Lung Cancer 2016, 17, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, K.; Mani, C.; Barnett, R.; Nalluri, S.; Bachaboina, L.; Rocconi, R.P.; Athar, M.; Owen, L.B.; Palle, K. Gli1 protein regulates the S-phase checkpoint in tumor cells via Bid protein, and its inhibition sensitizes to DNA topoisomerase 1 inhibitors. J. Biol. Chem. 2014, 289, 31513–31525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, R.; Gu, D.; Zhang, X.; Yu, B.; Liu, B.; Xie, J. The role of GLI1 for 5-Fu resistance in colorectal cancer. Cell Biosci. 2017, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Newbold, A.; Gould, C.M.; Luu, J.; Trapani, J.A.; Matthews, G.M.; Simpson, K.J.; Johnstone, R.W. A genome scale RNAi screen identifies GLI1 as a novel gene regulating vorinostat sensitivity. Cell Death Differ. 2016, 23, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Gu, D.; Zhang, X.; Li, J.; Liu, B.; Xie, J. GLI1-mediated regulation of side population is responsible for drug resistance in gastric cancer. Oncotarget 2017, 8, 27412–27427. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Gu, D.; Zhang, X.; Liu, B.; Xie, J. The role of GLI2-ABCG2 signaling axis for 5Fu resistance in gastric cancer. J. Genet. Genom. 2017, 44, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Gong, A.; Yang, H.; George, S.K.; Jiao, Z.; Huang, H.; Jiang, X.; Zhang, Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett. 2015, 369, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-M.; Lee, T.E.; Syed, S.A.; Burgoyne, A.M.; Leonard, S.Y.; Gao, F.; Chan, J.C.; Shi, E.; Chmielecki, J.; Morosini, D.; et al. Hedgehog pathway dysregulation contributes to the pathogenesis of human gastrointestinal stromal tumors via GLI-mediated activation of KIT expression. Oncotarget 2016, 7, 78226–78241. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Wang, X.; Baladandayuthapani, V.; Liu, B.; Shiozaki, H.; Shimodaira, Y.; Lin, Q.; Elimova, E.; Hofstetter, W.L.; Swisher, S.G.; et al. Nuclear expression of Gli-1 is predictive of pathologic complete response to chemoradiation in trimodality treated oesophageal cancer patients. Br. J. Cancer 2017, 117, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhou, X.-T.; Zou, H.-Y.; Wu, J. Hedgehog signaling pathway affects the sensitivity of hepatoma cells to drug therapy through the ABCC1 transporter. Lab. Investig. 2017, 97, 819–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, J.R.; Morgan, J.T.; Ioele, S.A.; Gleghorn, J.P.; Sims-Mourtada, J.; Day, E.S. Investigating the role of Hedgehog/GLI1 signaling in glioblastoma cell response to temozolomide. Oncotarget 2018, 9, 27000–27015. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, D.; Qian, Z.; Cui, D.; Gao, L.; Lou, M. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, J.; Miyake, H.; Fujisawa, M. GLI2 expression levels in radical nephrectomy specimens as a predictor of disease progression in patients with metastatic clear cell renal cell carcinoma following treatment with sunitinib. Mol. Clin. Oncol. 2016, 5, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Yan, L.; Lu, C.; Zhang, C.; Zhu, F.; Yang, J.; Jing, H.; Zhang, Y.; Qiao, J.; Guo, H. Activation of hedgehog signaling and its association with cisplatin resistance in ovarian epithelial tumors. Oncol. Lett. 2018, 15, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, F.; Wang, Y.; Zhang, Y.; Meng, L.; Chi, Y. Rab23 promotes the cisplatin resistance of ovarian cancer via the Shh-Gli-ABCG2 signaling pathway. Oncol. Lett. 2018, 15, 5155–5160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Eagles, J.; Keysar, S.B.; Wang, G.; Glogowska, M.J.; Altunbas, C.; Anderson, R.T.; Le, P.N.; Morton, J.J.; Frederick, B.; et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014, 74, 7024–7036. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xu, A.; Xu, J.; Huang, H.; Chen, L.; Su, Y.; Zhang, L.; Li, J.; Fan, F.; Deng, J.; et al. MicroRNA-324-5p regulates stemness, pathogenesis and sensitivity to bortezomib in multiple myeloma cells by targeting hedgehog signaling. Int. J. Cancer 2018, 142, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Y.; Lu, L.; Huang, S.; Ding, Y.; Zhang, Y.; Guo, Q.; Li, Z.; Zhao, L. Oroxyloside A Overcomes Bone Marrow Microenvironment-Mediated Chronic Myelogenous Leukemia Resistance to Imatinib via Suppressing Hedgehog Pathway. Front. Pharmacol. 2017, 8, 526. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, F.; Zhu, Q.; Ding, B.; Zhong, Q.; Huang, K.; Jiang, X.; Wang, Z.; Yin, C.; Zhu, Y.; et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget 2016, 7, 33004–33015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M. ABC transporters as molecular effectors of pancreatic oncogenic pathways: The Hedgehog-GLI model. J. Gastrointest. Cancer 2010, 41, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Amable, L.; Fain, J.; Gavin, E.; Reed, E. Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol. Rep. 2014, 32, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
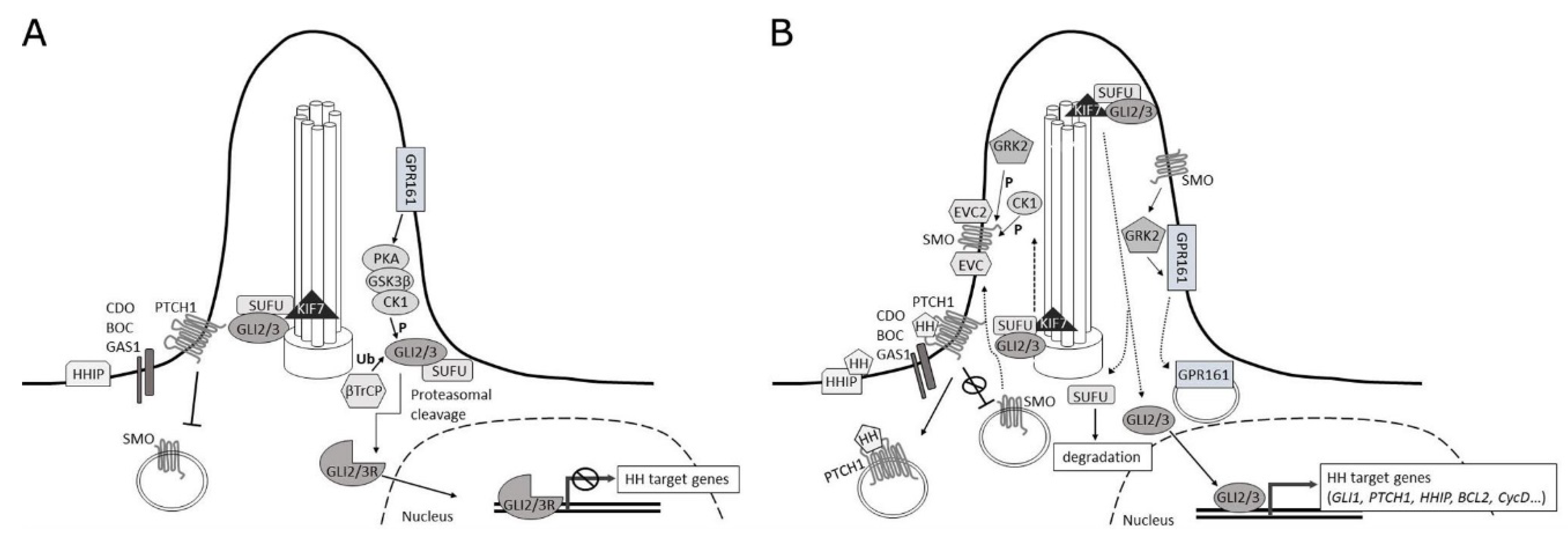{kind=link}
{kind=link}
| Syndrome/Condition | MIM ID# | Hand Defects | Feet Defects | Cranial Malformations | Intellectual Disability | Other | Reference |
|---|---|---|---|---|---|---|---|
| Greig cephalopolysyndactyly syndrome | 175700 | postaxial polydactyly, cutaneous syndactyly | preaxial polydactyly, cutaneous syndactyly | hypertelorism, macrocephaly with frontal bossing | sometimes | central nervous system (CNS) anomalies, hernias | [93] |
| Greig cephalopolysyndactyly contiguous gene deletion syndrome | 175700 | postaxial polydactyly, cutaneous syndactyly | preaxial polydactyly, cutaneous syndactyly | hypertelorism, macrocephaly, hydrocephalus | often | seizures, ophthalmologic findings | [57,92] |
| Acrocallosal Syndrome | 200990 | preaxial and postaxial polydactyly, cutaneous syndactyly | preaxial polydactyly | macrocephaly, widely spaced eyes, absence of the corpus callosum, hydrocephalus | yes | seizures, interhemispheric cysts, umbilical hernia | [95,96] |
| Pallister-Hall syndrome | 146510 | central polydactyly, postaxial polydactyly | none | bifid epiglottis | no | hypothalamic hamartoma, imperforate anus, renal abnormalities, genital defects | [57,97] |
| Inhibitor | Mode of Action | Model/Samples | Effect | Reference |
|---|---|---|---|---|
| Compounds affecting GLI activity | ||||
| GANT58 | GLI antagonist | Acute T lymphocytic leukemia | Decreased cell viability, G1/S accumulation | [216] |
| GANT61 | Inhibition of GLI1/DNA and GLI2/DNA binding | Colon carcinoma | Cytotoxicity, induction of DNA damage, G1/S accumulation | [202] |
| Large cell neuroendocrine carcinoma of the lung | Inhibition of cell growth, sensitivity to cisplatin | [217] | ||
| Prostate cancer | Decreased cell viability, induction of apoptosis, downregulation of stem cell markers | [214,218] | ||
| Acute myeloid leukemia | Inhibition of cell growth and colony formation, induction of apoptosis | [215,219] | ||
| Rhabdomyosarcoma | Inhibition of xenograft growth, induction of apoptosis, downregulation of EMT markers | [220] | ||
| Neuroblastoma | Decreased cell viability, induction of apoptosis, induction of autophagy, inhibition of xenograft growth | [201,221] | ||
| Ewing’s sarcoma family tumor | Cytotoxicity, induction of apoptosis | [222] | ||
| Ovarian cancer | Reduced migration and invasion, inhibition of xenograft growth | [223] | ||
| Squamous lung cancer | Inhibition of cell growth, induction of apoptosis, inhibition of xenograft growth | [210] | ||
| Pancreatic cancer stem cells | Decreased cell viability, reduced migration, invasion and spheroid formation, induction of apoptosis, inhibition of xenograft growth | [204] | ||
| T-cell lymphoma | Decreased cell viability, induction of apoptosis | [224] | ||
| Mesothelioma | Cytotoxicity, induction of apoptosis, G1 accumulation | [203] | ||
| Biliary tract tumors | Cytotoxicity, induction of apoptosis | [212] | ||
| Cervical cancer | Reduction of proliferation and survival, induction of apoptosis, generation of ROS | [225] | ||
| Melanoma | Decreased cell viability, induction of apoptosis, reduced colony forming | [226] | ||
| arsenic trioxide (ATO) | Inhibition of GLI1/GLI2 activity | Pancreatic cancer stem cells | Inhibition of cell growth, induction of apoptosis, reduced migration | [227] |
| Medulloblastoma | Cytotoxicity, inhibition of xenograft growth | [228,229] | ||
| Osteosarcoma | Inhibition of cell growth, reduced colony forming, induction of apoptosis, inhibition of xenograft growth | [230,231] | ||
| Prostate | Inhibition of cell growth | [232] | ||
| Rhabdoid tumors | Cytotoxicity, induction of apoptosis, inhibition of xenograft growth | [233] | ||
| cynanbungeigenin C and D (CBC and CBD) | Gli1 antagonists | Medulloblastoma | Inhibition of allograft growth | [234] |
| gedunin | GLI inhibition | Pancreatic cancer | Inhibition of cell growth, induction of apoptosis, reduced migration, downregulation of EMT markers, inhibition of xenograft growth | [235] |
| GLI-I | GLI inhibitor | Malignant pleural mesothelioma | Cytotoxicity, induction of apoptosis, inhibition of xenograft growth | [236] |
| glabrescione B | Inhibition of GLI1/DNA binding | Medulloblastoma, basal cell carcinoma | Inhibition of cell growth, reduced spheroid formation, induction of apoptosis, inhibition of allograft growth | [237] |
| physalin H | GLI1-DNA complex formation inhibition | Pancreatic and prostate cancer | Cytotoxicity | [238] |
| vitretrifolin D | Inhibition of GLI1-DNA binding | Prostate and pancreatic cancer | Cytotoxicity | [239] |
| solasonine | GLI-mediated transcription | SHH-LIGHT2 reporter cells | downregulation of GLI1 and PTCH1 expression | [240] |
| Compounds affecting GLI stability and trafficking | ||||
| HPI-1-4 | Different effects on GLI stability and trafficking | Breast cancer | Inhibition of cell growth, induction of apoptosis, reduced cancer stem cell population, reduced migration | [241,242] |
| aspirin | Inhibits GLI1 translocation to nucleus | Glioma | Cytotoxicity, induction of apoptosis, reduced migration and invasion, downregulation of EMT markers, | [243] |
| Compounds affecting GLI expression | ||||
| genistein | GLI1 expression | Gastric cancer | Downregulation of stem cell markers, reduced invasive capacity | [244] |
| Breast cancer | Inhibition of cell growth, reduced colony forming, induction of apoptosis, inhibition of xenograft growth | [245] | ||
| Quinolone-1-(2H)-ones | GLI expression | SHH-LIGHT2 reporter cells | Downregulation of GLI1 and PTCH1 expression | [246] |
| compound 29a | GLI protein expression | Medulloblastoma | Inhibition of allograft growth | [247] |
| Compounds affecting protein regulators of GLI activity | ||||
| imiquimod | PKA-mediated GLI phosphorylation | Murine asocellular carcinoma, human medulloblastoma | Downregulation of GLI1 and HHIP expression, GLI3 processing | [248] |
| forskolin | PKA activation | Pediatric tumors | Inhibition of cell growth, induction of apoptosis | [249] |
| perifosine | inhibition of GLI1 via AKT/PI3K | Pancreatic cancer | PTCH1 downregulation, cytotoxicity, senzitization to gemcitabine | [250] |
| Acute T cell leukemia | Cytotoxycity | [216] | ||
| nanoquinacrine | activation of GSK3B | Oral cancer stem cells | Inhibition of cell growth, induction of apoptosis | [251] |
| Epigenetic regulation of GLI activity | ||||
| JQ1 | Inhibition of BET bromodomain | medulloblastoma, basocellular carcinoma | Inhibition of cell growth, induction of apoptosis, inhibition of allograft growth | [252] |
| I-BET151 | Inhibition of BET bromodomain | medulloblastoma | Downregulation of GLI1, inhibition of allograft growth | [253] |
| Combination Therapy | Molecular Targets | Model | Effect | Reference |
|---|---|---|---|---|
| ATO + radiotherapy | GLI + proliferation | High grade neuroepithelial tumor of the central nervous system (primary culture and patient) | Clinical remission for 6 months | [260] |
| ATO + itraconazole | GLI + SMO | Patients with metastatic basal cell carcinoma | Best overall response was stable disease in 3/5 patients | [261] |
| GANT61 + obatoclax | GLI + BCL2 | Melanoma cells | Decreased cell viability, increased apoptosis | [226] |
| GANT61 + sunitinib + PF-04691502 | GLI + FLT3 + PI3K | Acute myeloid leukemia | Stronger anti-leukemic effects in vivo, prolonged survival of mice | [262] |
| GANT61 + antiestrogens | GLI + estrogen | ER+ breast cancer cell lines | Decreased cell growth | [263] |
| GANT61 + metformin | GLI + gluconeogenesis | Prostate cancer cells and xenografts | Decreased cell growth, enhanced radiation response | [264] |
| GANT61 + paclitaxel | GLI + spindle inhibition | ER+ and triple negative breast cancer | Decreased cell growth in TNBC | [265] |
| GANT61 + temozolomide | GLI + alkylation/methylation of DNA | Glioma cell lines | Decreased cell growth, increased apoptosis, GANT61 sensitizes cells to TMZ | [266,267] |
| GANT61 + rapamycin | GLI + mTOR | Pancreatic cancer cell lines and xenografts | Reduced sphere formation and cell viability | [268] |
| curcumin + resveratrol | HH-GLI signaling | Breast cancer cell lines and xenografts | Induction of apoptosis, 10-fold lower IC50 doses in combination compared to individual treatments | [269] |
| ATO + LY294002 | GLI + PI3K | Colon carcinoma cells | Decreased proliferation, synergistic effect | [270] |
| GANT58 + perifosine | GLI + AKT/PI3K | Acute T cell leukemia | Synergistic cytotoxic effect | [216] |
| GANT61 + rapamycin | GLI + mTOR | Myeloid leukemia | Growth arrest and apoptosis, synergistic effect | [271] |
| GANT61 + itraconazole | GLI + antifungal | Breast cancer cell lines | Synergistically enhanced cytotoxicity | [272] |
| GANT61 + PI103 | GLI + PI3K/mTOR | Rhabdomyosarcoma cell lines and xenografts | Synergistic apoptosis induction and tumor growth reduction | [273] |
| GANT61 + trastuzumab + BEZ235 | GLI + ErbB2-PI3K-mTORC1 | Esophageal carcinoma | Significantly stronger tumor reduction then individual treatments | [274] |
| GANT61 + irradiated riboflavin (iRF) | GLI + photosensitizer | Melanoma cells and mouse model | Potentiates the antiproliferative effect of iRF | [275] |
| GANT61 + cisplatin/doxorubicin/Irinotecan/vincristine | GLI + standard chemotherapy | Neuroblastoma | Synergistic (doxorubicin or vincristine) or additive effects (cisplatin or irinotecan) | [201] |
| GANT61 + cisplatin | GLI + standard chemotherapy | Biliary tract cancer | Synergistic effect | [212] |
| GANT61 + rapamycin/temsirolimus | GLI + mTOR | Rhabdomyosarcoma | Reduced survival compared to individual treatments | [220] |
| ATO + cis-platin/ifosfamide/doxorubicin + vismodegib | GLI + standard chemotherapy + SMO | Osteosarcoma | Synergistic effect, inhibition of tumor growth in vivo | [231] |
| nanoHHI + gemcitabine | GLI + nucleoside synthesis | Pancreatic cancer | Inhibition of tumor growth in vivo | [257] |
| ATO + cyclopamine | GLI + SMO | Prostate cancer | Synergistic effect, inhibition of tumor growth in vivo | [232] |
| ATO + LY294002 | GLI + PI3K | Colon cancer cells | Synergistic effect | [270] |
| ATO + gemcitabine | GLI + nucleoside synthesis | Pancreatic cancer cell lines and xenografts | Synergistic effect, inhibition of tumor growth in vivo | [227] |
| Tumor Type | Resistant to: | Resistance Mechanism | Reference |
|---|---|---|---|
| Lung cancer | Staurosporine | GLI1-mediated upregulation of NDRG1 and downregulation of c-MYC and N-MYC | [276] |
| Gefitinib | Upregulation of SHH, SMO and GLI1 (reversible by sulforaphane) | [277] | |
| Platinum and gefitinib/erlotinib | MEOX-2-dependent GLI1 upregulation | [278] | |
| Platinum | Hedgehog pathway activation | [279] | |
| EGFR inhibitors | GLI1-mediated upregulation of SOX2, induction of EMT | [105,280] | |
| Lung and colorectal cancer | Topoisomerase inhibitors | GLI1-mediated upregulation of BID | [281] |
| Colorectal cancer | 5-Fluorouracil | Upregulation of GLI1 and GLI2 and their targets | [282] |
| Vorinostat | GLI1-mediated upregulation of BCL2L1 | [283] | |
| Gastrointestinal cancer | 5-Fluorouracil, cisplatin | GLI2-mediated upregulation of ABCG2 transporter | [284,285] |
| Doxorubicin | GLI2-mediated upregulation of ABCG2 transporter | [286] | |
| Imatinib | GLI-mediated upregulation of KIT | [287] | |
| Esophageal cancer | Chemoradiation | Nuclear localization of GLI1 | [288] |
| Hepatocellular cancer | Sorafenib | GLI2-mediated upregulation of ABCC1 transporter | [289] |
| Glioblastoma | Temozolomide | GLI1-mediated upregulation of MGMT | [290,291] |
| Clear cell renal carcinoma | Sunitinib | GLI2 overexpression | [292] |
| Ovarian cancer | Cisplatin | Rab23-mediated upregulation of ABCG2 through GLI1 | [293,294] |
| Basal cell carcinoma | Gdc-619 | SRF-mediated upregulation of GLI1 | [295] |
| Head and neck squamous cancer | Radioresistance | mTOR/SK6-mediated upregulation of GLI1 | [296] |
| Multiple myeloma | Lenalidomide | ADAR1-dependent RNA editing of GLI1 | [144] |
| Bortezomib | Downregulation of mir-324-5p leading to upregulation of SMO and GLI1 | [297] | |
| Chronic myeloid leukemia | Imatinib | GLI1-mediated upregulation of BCR-ABL, p-Akt, Bcl-xl and survivin (reversible by oroxyloside A) | [298] |
| Acute myeloid leukemia | Radiation | Activation of the GLI1/PI3K/AKT/NFKB pathway | [299] |
| Ribavirin/cytarabine | GLI1-mediated upregulation of UDP-glucuronosyl transferase enzymes | [300] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabol, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2562. https://doi.org/10.3390/ijms19092562
Sabol M, Trnski D, Musani V, Ozretić P, Levanat S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. International Journal of Molecular Sciences. 2018; 19(9):2562. https://doi.org/10.3390/ijms19092562
Chicago/Turabian StyleSabol, Maja, Diana Trnski, Vesna Musani, Petar Ozretić, and Sonja Levanat. 2018. "Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets" International Journal of Molecular Sciences 19, no. 9: 2562. https://doi.org/10.3390/ijms19092562