Structural Prediction of the Dimeric Form of the Mammalian Translocator Membrane Protein TSPO: A Key Target for Brain Diagnostics

 , , , , , and
, , , , , and
Abstract
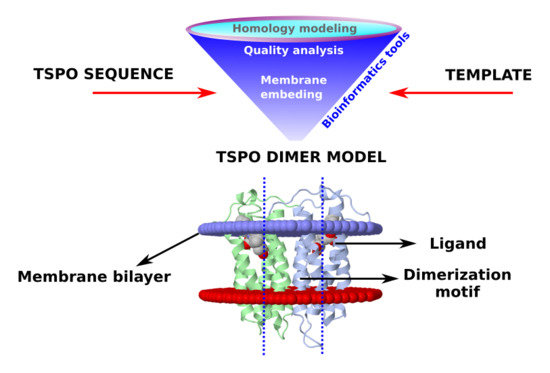
:
1. Introduction
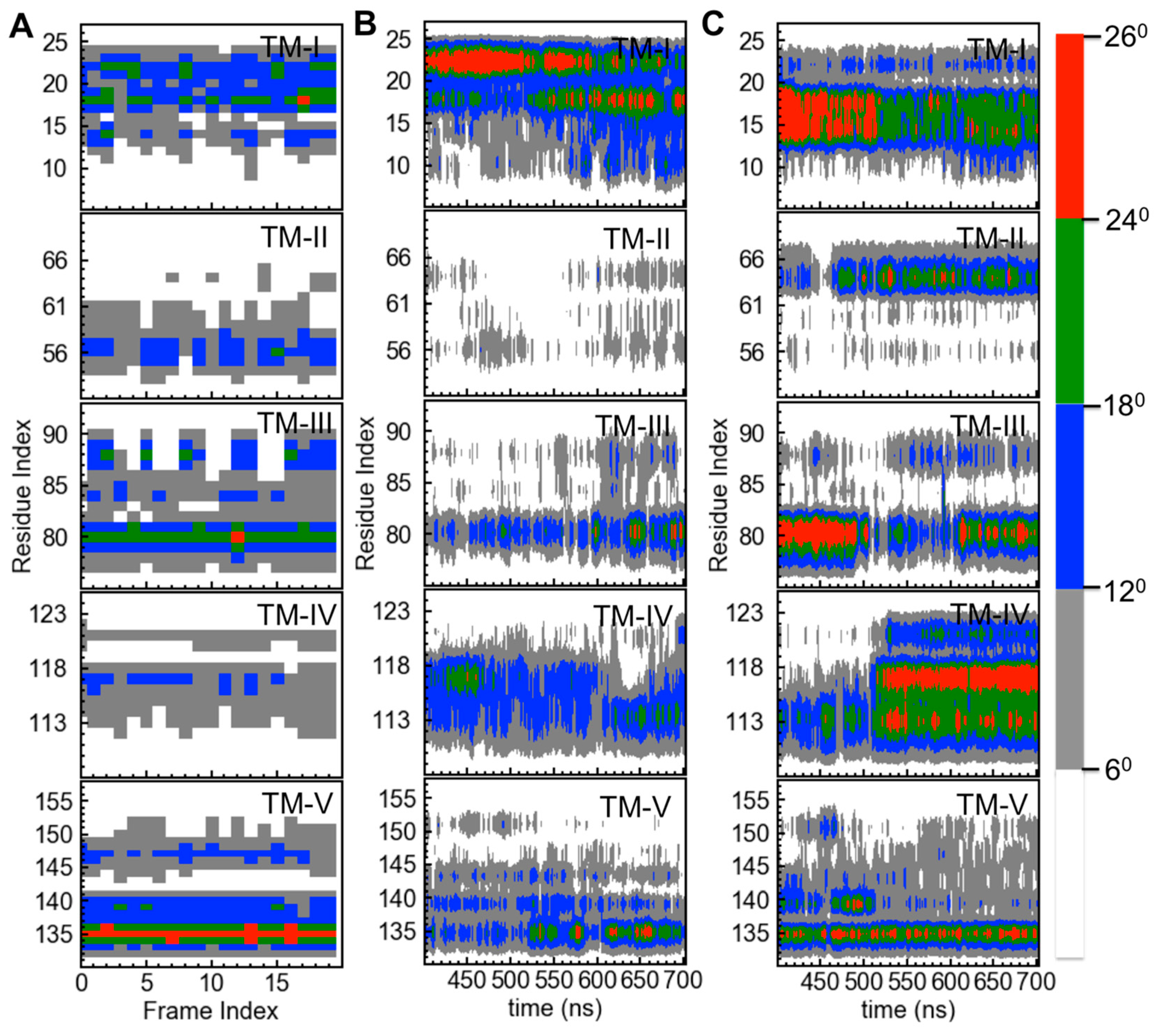
2. Results
Analysis of the Templates for mTSPO_NMR and for mTSPO_Rs
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analyses
4.2. MD Simulations of mTSPO_NMR_monomer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TSPO | Translocator membrane protein |
| PET | Positron emission tomography |
| RsTSPO | Rhodobacter Sphaeroides |
| BcTSPO | Bacillus Cereus |
| mTSPO_NMR_monomer | Monomer of mTSPO solved by solution NMR experiment, PDBiD: 2MGY |
| mTSPO_NMR | Dimer model of mTSPO. The prediction is based on mTSPO_NMR_monomer structure |
| mTSPO_Rs | Dimer model of mTSPO. The prediction is based on the RsTSPO structure |
| MD | Molecular dynamics |
| bb-RMSD | Root-mean square deviation of backbone atoms (N, Cα, C atoms) |
| RMSF | Root-mean square fluctuation |
References
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.-J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.-R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Beatriz, C.; Leo, V.; Moshe, G. Role of mitochondrial translocator protein (18 kDa) on mitochondrial-related cell death processes. Recent Pat. Endocr. Metab. Immune Drug Discov. 2013, 7, 86–101. [Google Scholar] [CrossRef]
- Papadopoulos, V. On the role of the translocator protein (18-kDa) TSPO in steroid hormone biosynthesis. Endocrinology 2014, 155, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Auta, J.; Guidotti, A.; Korneyev, A.; Romeo, E. The pharmacology of neurosteroidogenesis. J. Steroid Biochem. Mol. Biol. 1994, 49, 385–389. [Google Scholar] [CrossRef]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin. Transl. Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, A.; Palazzo, C.; de Candia, M.; Lasorsa, F.M.; Trapani, G. Targeting of the translocator protein 18 kDa (TSPO): A valuable approach for nuclear and optical imaging of activated microglia. Bioconjugate Chem. 2013, 24, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Dolle, F.; Luus, C.; Reynolds, A.; Kassiou, M. Radiolabelled molecules for imaging the translocator protein (18 kDa) using positron emission tomography. Curr. Med. Chem. 2009, 16, 2899–2923. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Pae, A.N. Translocator protein (TSPO) ligands for the diagnosis or treatment of neurodegenerative diseases: A patent review (2010–2015; part 1). Expert Opin. Ther. Pat. 2016, 26, 1325–1351. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Pae, A.N. Translocator protein (TSPO) ligands for the diagnosis or treatment of neurodegenerative diseases: A patent review (2010–2015; part 2). Expert Opin. Ther. Pat. 2016, 26, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Kodo, T.; Ishiyama, T.; Kakuyama, H.; Nishikawa, H.; Baba, S.; Masumoto, S. Design, synthesis and structure-activity relationships of novel benzoxazolone derivatives as 18kDa translocator protein (TSPO) ligands. Bioorg. Med. Chem. 2012, 20, 5568–5582. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Veenman, L.; Shterenberg, A.; Singh, S.; Masarwa, A.; Dutta, B.; Island, B.; Tsoglin, E.; Levin, E.; Leschiner, S.; et al. Quinazoline-based tricyclic compounds that regulate programmed cell death, induce neuronal differentiation, and are curative in animal models for excitotoxicity and hereditary brain disease. Cell Death Discov. 2015, 1, 15027. [Google Scholar] [CrossRef] [PubMed]
- Simon-O’Brien, E.; Gauthier, D.; Riban, V.; Verleye, M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J. Neuroinflammation 2016, 13, 203. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Cavallini, C.; Da Pozzo, E.; Taliani, S.; Da Settimo, F.; Martini, C. The anxiolytic etifoxine binds to TSPO Ro5-4864 binding site with long residence time showing a high neurosteroidogenic activity. ACS Chem. Neurosci. 2017, 8, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Veenman, L.; Singh, S.; Ouyang, F.; Liang, J.; Huang, W.; Marek, I.; Zeng, J.; Gavish, M. 2-Cl-MGV-1 ameliorates apoptosis in the thalamus and hippocampus and cognitive deficits after cortical infarct in rats. Stroke 2017, 48, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Delavoie, F.; Li, H.; Hardwick, M.; Robert, J.-C.; Giatzakis, C.; Péranzi, G.; Yao, Z.-X.; Maccario, J.; Lacapère, J.-J.; Papadopoulos, V. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: Functional significance in drug ligand and cholesterol binding. Biochemistry 2003, 42, 4506–4519. [Google Scholar] [CrossRef] [PubMed]
- Boujrad, N.; Vidic, B.; Papadopoulos, V. Acute action of choriogonadotropin on Leydig tumor cells: Changes in the topography of the mitochondrial peripheral-type benzodiazepine receptor. Endocrinology 1996, 137, 5727–5730. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2017, 30, e12500. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Jaipuria, G.; Leonov, A.; Giller, K.; Vasa, S.K.; Jaremko, Ł.; Jaremko, M.; Linser, R.; Becker, S.; Zweckstetter, M. Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure. Nat. Commun. 2017, 8, 14893. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y.-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosig, B.; Langosch, D. The dimerization motif of the glycophorin a transmembrane segment in membranes: Importance of glycine residues. Protein Sci. 2008, 7, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Gerstein, M.; Engelman, D.M. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol. 2000, 296, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Ubarretxena-Belandia, I.; Engelman, D.M. The Cα—H···O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 9056. [Google Scholar] [CrossRef] [PubMed]
- Doura, A.K.; Fleming, K.G. Complex interactions at the helix-helix interface stabilize the glycophorin a transmembrane dimer. J. Mol. Biol. 2004, 343, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Liu, N.; Kuhn, L.A.; Garavito, R.M.; Ferguson-Miller, S. Translocator protein 18 kDa (TSPO): An old protein with new functions? Biochemistry 2016, 55, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, L.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Lacapère, J.-J.; Delavoie, F.; Li, H.; Péranzi, G.; Maccario, J.; Papadopoulos, V.; Vidic, B. Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res. Commun. 2001, 284, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Scarf, A.M.; Kassiou, M. The translocator protein. J. Nucl. Med. 2011, 52, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xia, Y.; Meiler, J.; Ferguson-Miller, S. Characterization and modeling of the oligomeric state and ligand binding behavior of purified translocator protein 18 kDa from rhodobacter sphaeroides. Biochemistry 2013, 52, 5884–5899. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, j.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kalathur, R.C.; Liu, Q.; Kloss, B.; Bruni, R.; Ginter, C.; Kloppmann, E.; Rost, B.; Hendrickson, W.A. Structure and activity of tryptophan-rich TSPO proteins. Science 2015, 347, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, M.G.; Carlson, H.A. APBS Plugin for PyMOL; University of Michigan: Ann Arbor, MI, USA, 2006. [Google Scholar]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sotriffer, C.A. Accounting for induced-fit effects in docking: What is possible and what is not? Curr. Top. Med. Chem. 2011, 11, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.L.C. Glide; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2017. [Google Scholar]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, J.; Barrantes, F. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.F.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, A.C.; Chavent, M.; Sansom, M.S. Bendix: Intuitive helix geometry analysis and abstraction. Bioinformatics 2012, 28, 2193–2194. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef] [PubMed]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14532. [Google Scholar] [CrossRef] [PubMed]
- Kleiger, G.; Grothe, R.; Mallick, P.; Eisenberg, D. GXXXG and AXXXA: Common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry 2002, 41, 5990–5997. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol. 2017, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Protein Structure Prediction; Kihara, D., Ed.; Springer New York: New York, NY, USA, 2014; pp. 1–15. [Google Scholar]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2009, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Bino, J.; Sali, A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003, 31, 3982–3992. [Google Scholar] [CrossRef] [Green Version]
- Baker, F.N.; Porollo, A. CoeViz: A web-based tool for coevolution analysis of protein residues. BMC Bioinform. 2016, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Di Nardo, A.A.; Davidson, A.R. Analysis of covariation in an SH3 domain sequence alignment: Applications in tertiary contact prediction and the design of compensating hydrophobic core substitutions. J. Mol. Biol. 2000, 303, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. Anisotropic solvent model of the lipid bilayer. 2. energetics of insertion of small molecules, peptides, and proteins in membranes. J. Chem. Inf. Model. 2011, 51, 930–946. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.7.4.5; Edu Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.M.; Lyubartsev, A.P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.M.; Lyubartsev, A.P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The resp model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.M.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Hunenberger, P. Thermostat algorithms for molecular dynamics simulations. Adv. Polym. Sci. 2005, 173, 105–147. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Zimmermann, K. Oral: All purpose molecular mechanics simulator and energy minimizer. J. Comput. Chem. 1991, 12, 310–319. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Paramo, T.; East, A.; Garzón, D.; Ulmschneider, M.B.; Bond, P.J. Efficient characterization of protein cavities within molecular simulation trajectories: Trj_cavity. J. Chem. Theory Comput. 2014, 10, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Rossetti, G.; Carloni, P. Local fluctuations and conformational transitions in proteins. J. Chem. Theory Comput. 2012, 8, 4775–4785. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Inferred by Experiment | mTSPO_NMR | mTSPO_Rs | ||
|---|---|---|---|---|---|
| Binding Cavity | G19, A23, V26, R27, H43, R46, L49, A50, I52, W53, W95, W107, A110, D111, L114, W143, A147, L150, N151 | Subunit A | Subunit B | Subunit A | Subunit B |
| G18, G19, F20, G22, A23, V26, R27, G30, L31, K39, P40, S41, H43, P44, P45, R46, L49, A50, I52, W53, W93, W95, W107, A108, A110, D111, L114, W143, F146, A147, T148, L150, N151 | G18, G19, F20, G22, A23, V26, R27, G30, L31, K39, P40, S41, H43, P44, P45, R46, L49, A50, I52, W53, W93, W95, W107, A108, A110, D111, L114, W143, F146, A147, T148, L150, N151 | P15, G18, G19, M21, G22, A23, F25, V26, R27, G28, E29, Y34, K39, H43, P44, R46, L49, A50, W53, G54, Y57, N92, W93, W95, P96, F99, F100, L112, W143, F146, A147, T148, L150, N151, V154 | G18, M21, G22, A23, F25, Y34, H43, P44, R46, L49, A50, W53, L56, Y57, N92, W93, A9, W95, P96, P97, F99, F100, L112, V115, Y140, L141, W143, A147, L150 | ||
| Dimer Interface | V80, G83, Q88, N92, W93, W95, I98, F100, G101, A102, D111, V118 | F74, T75, E76, D77, M79, V80, P81, G83, L84, T86, G87, Q88, A90, L91 | V6, P7, G10, L11, L13, V14, L17, G18, F20, M21, Y24 V26, R27(A) M79, V80, L82, G83, L84, Y85, T86, G87, A90, L91, W93, A94, P97, I98, A102, Q104, W107, A108, A110, D111, L114, V118, A121, A125 | ||
| Conserved Residues | LP-I: L37, P40, P44, P45, TM-II: W53, L56, G61, TM-III: N92, W95, F99, F100, TM-V: L136, P139, Y140, W143, A147, L150, N151 | ||||
| Evolutionary Coupling | P40, P45 coevolve with L150; P44, P45 with W95; W53 with L56, A147, L150; W95 with A147 and N151 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, J.; Guareschi, R.; Damre, M.; Cao, R.; Kless, A.; Neumaier, B.; Bauer, A.; Giorgetti, A.; Carloni, P.; Rossetti, G. Structural Prediction of the Dimeric Form of the Mammalian Translocator Membrane Protein TSPO: A Key Target for Brain Diagnostics. Int. J. Mol. Sci. 2018, 19, 2588. https://doi.org/10.3390/ijms19092588
Zeng J, Guareschi R, Damre M, Cao R, Kless A, Neumaier B, Bauer A, Giorgetti A, Carloni P, Rossetti G. Structural Prediction of the Dimeric Form of the Mammalian Translocator Membrane Protein TSPO: A Key Target for Brain Diagnostics. International Journal of Molecular Sciences. 2018; 19(9):2588. https://doi.org/10.3390/ijms19092588
Chicago/Turabian StyleZeng, Juan, Riccardo Guareschi, Mangesh Damre, Ruyin Cao, Achim Kless, Bernd Neumaier, Andreas Bauer, Alejandro Giorgetti, Paolo Carloni, and Giulia Rossetti. 2018. "Structural Prediction of the Dimeric Form of the Mammalian Translocator Membrane Protein TSPO: A Key Target for Brain Diagnostics" International Journal of Molecular Sciences 19, no. 9: 2588. https://doi.org/10.3390/ijms19092588
APA StyleZeng, J., Guareschi, R., Damre, M., Cao, R., Kless, A., Neumaier, B., Bauer, A., Giorgetti, A., Carloni, P., & Rossetti, G. (2018). Structural Prediction of the Dimeric Form of the Mammalian Translocator Membrane Protein TSPO: A Key Target for Brain Diagnostics. International Journal of Molecular Sciences, 19(9), 2588. https://doi.org/10.3390/ijms19092588