Experimental
Ceramic samples of La
1-xSr
xCo
1-yTM
yO
3 (TM=Fe, Ni) were synthesised having y = 0, 0.2,0.4,1.0 and x = 0, 0.2. Stoichiometric amounts of La
2O
3 (99.9%), SrCO
3(99.994%), CoC
2O
4 (and/or the respective Ni and Fe oxalates, all >99.99%) were ground together and annealed in air at 800°C for 24 hours, reground, and then heated further at 1000°C for 96 hours. After a further regrinding, the last annealing cycle was repeated. X-ray diffraction showed in general that only the perovskite ABO
3 phase was present, with a rhombohedral distortion which decreased with Sr doping level. For x=0, y=0.4 Fe, a mixed-phase rhombohedral and orthorhombic structure was formed, consistent with literature results [
5]. As might be expected from the high anealing temperatures used to synthesize these 'model catalysts', the samples were highly crystalline, as evidenced by the narrow reflections observed in XRD. Consistent with this, the BET surface areas of such ceramics are much lower than in a normal working catalyst material, typically around 0.5 m
2 g
-1, with small random fluctuations with doping level. These are insufficiently large to affect the catalytic trends presented below (e.g.
Table 2).
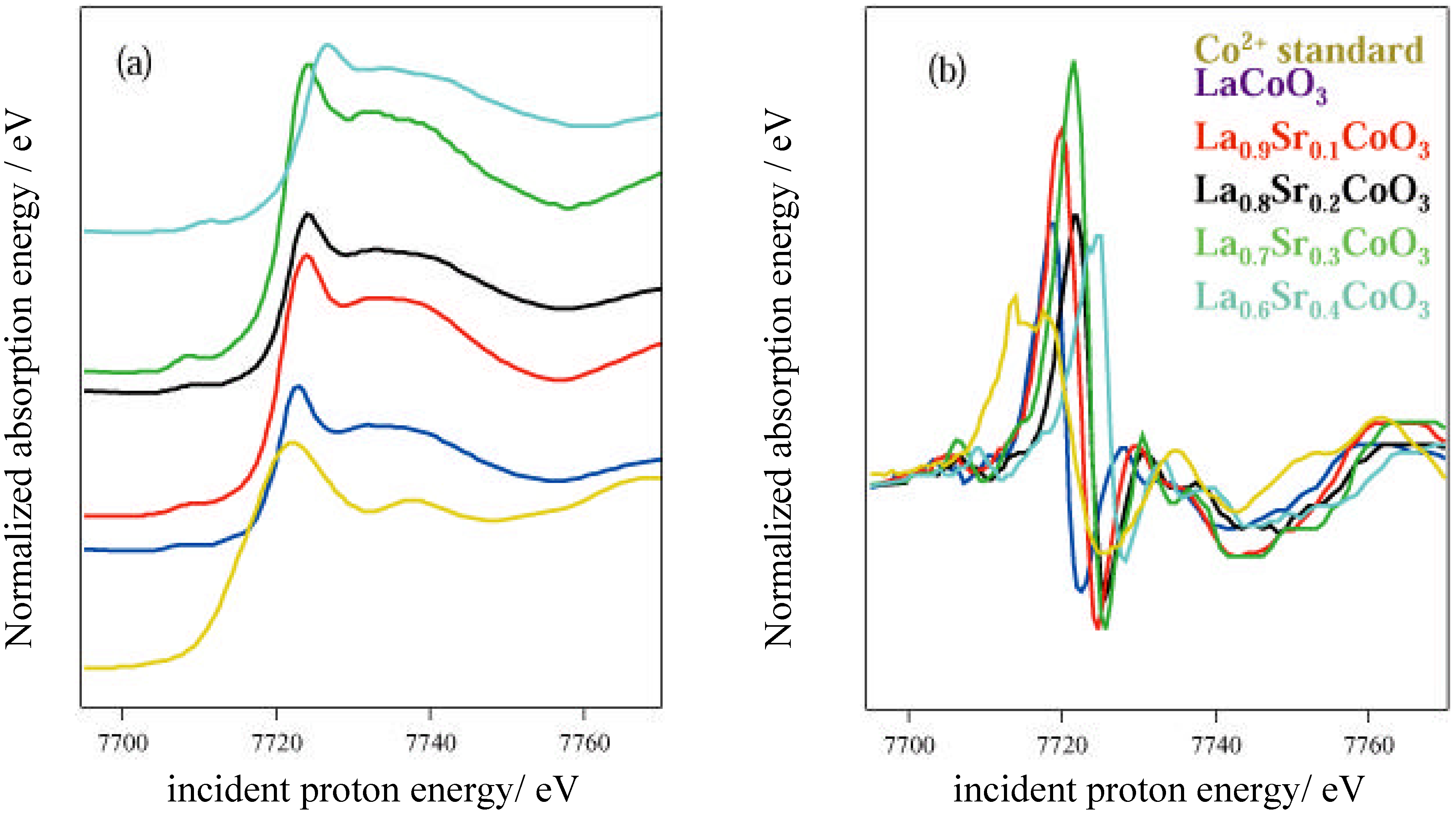
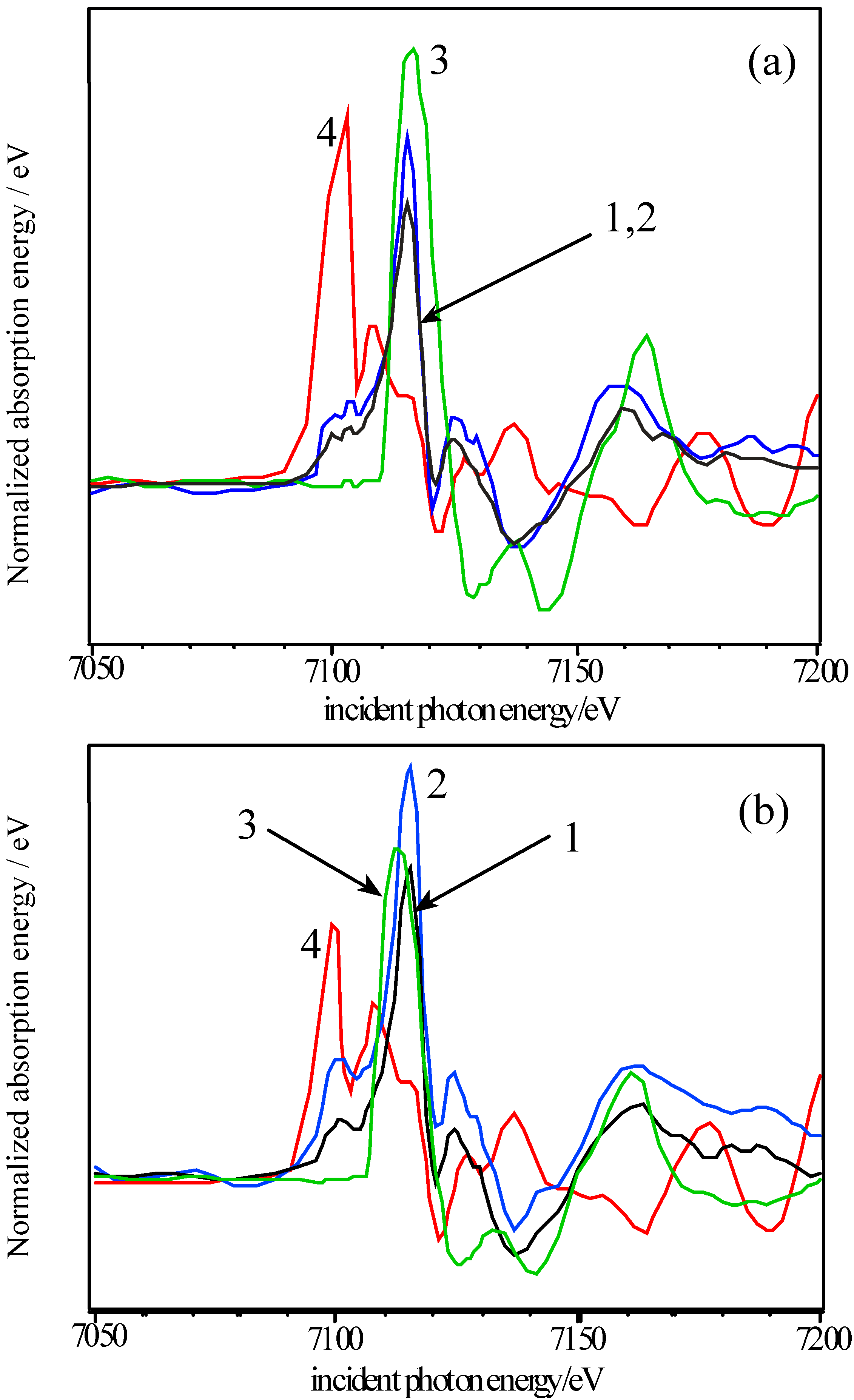
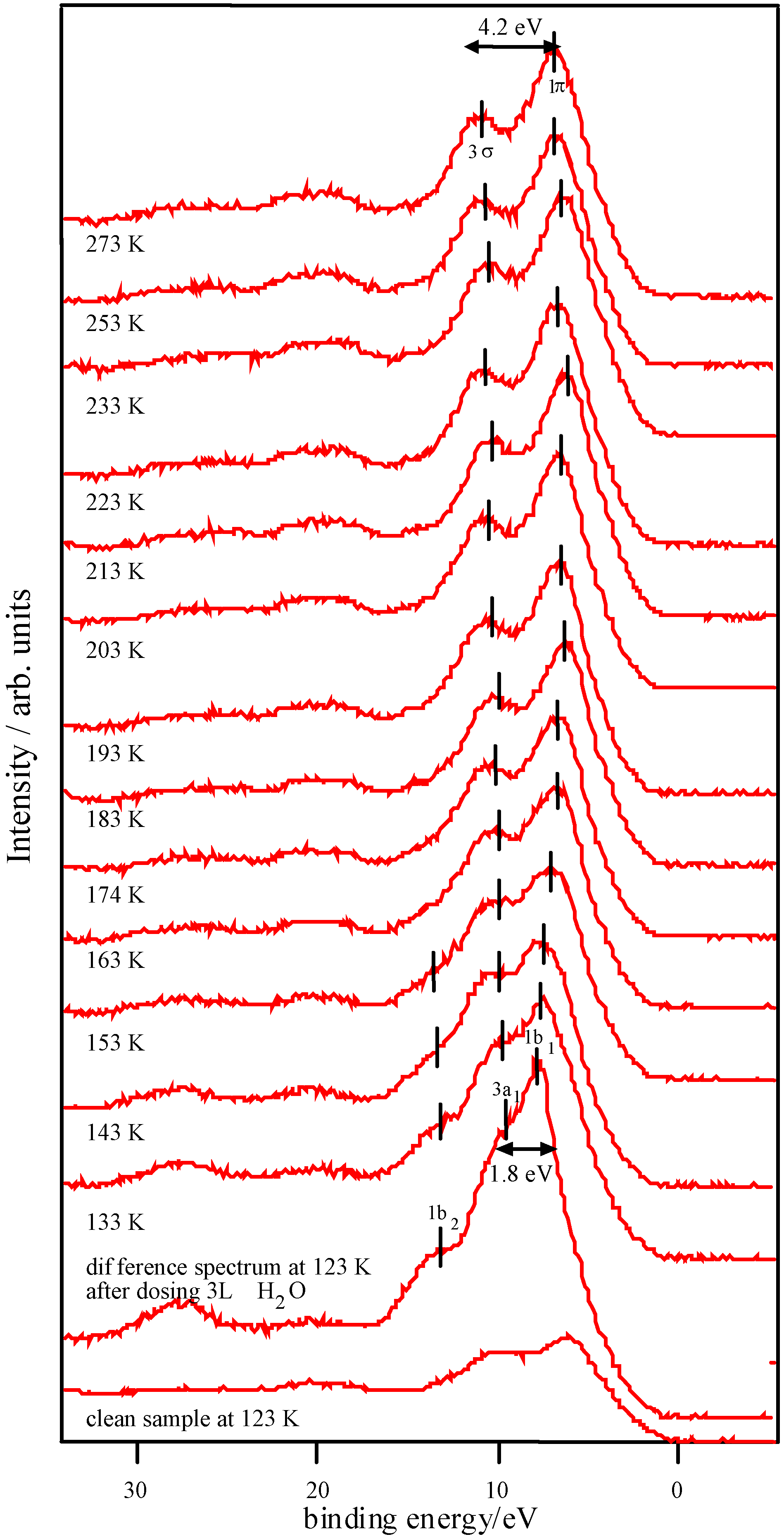
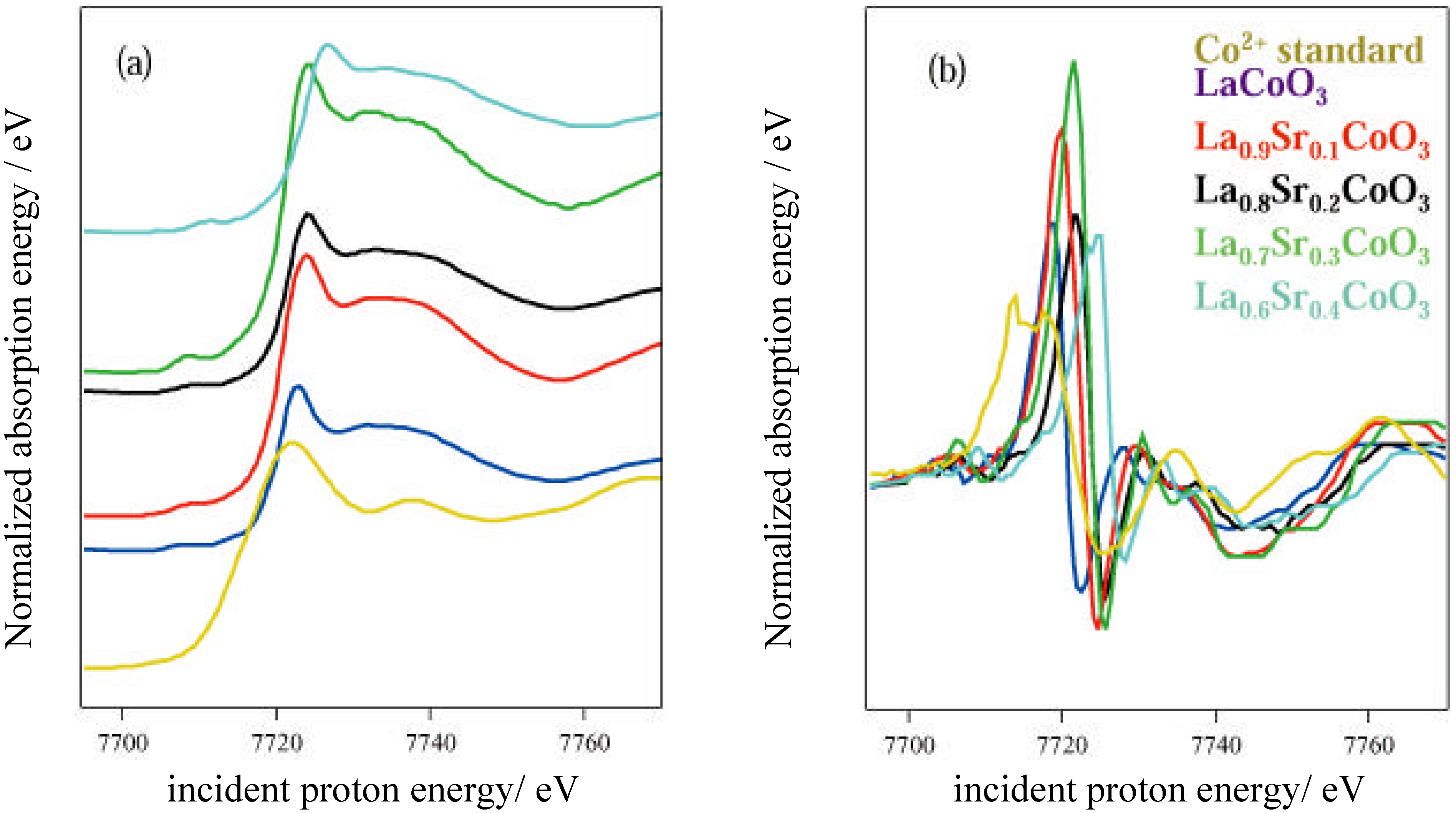
The activity of the samples in the decomposition of H2O2 was tested by measuring the volume of gas evolved from static reaction at 40 °C using a burette, as the peroxide decomposed to water and O2. Production of epoxide from crotyl alcohol over the catalyst in aqueous solution in the presence of H2O2 was measured using a flow reactor connected to a gas chromatography (GC) instrument (Perkin Elmer), calibrated using DMSO2. A 2M solution of epoxide with excess hydrogen peroxide was used, at a flow speed of 1 ml min-1 and a temperature of 40 °C. Atomic absorption (AA) spectroscopy using a Thermo Jarrell Ash Video 22E instrument was used to determine the metal content in the solutions following reaction. Photoemission and X-ray absorption measurements were carried out at the SRS, CLRC Daresbury Laboratory. Photoemission measurements used the spherical grating monochromator (15≤hν≤220 eV) on Station 4.1, with a Scienta 200 mm hemispherical analyzer. The position of the Fermi edge was determined by reference to the Mo sample holder. The surfaces of ceramic pellets of the catalysts were cleaned for UHV measurements by scraping using a diamond file in vacua of 5 x 10-11 – 10-10 torr. In order to investigate stability in water, a series of experiments were conducted in which the surfaces were dosed with freeze-degassed doubly-distilled water. This was admitted to the vacuum chamber via a leak valve, with the exposure in langmuirs (1 L = 10-6 torr s) estimated from the chamber ion gauge. The samples were initially cooled to around 125 K before dosing and then warmed to room temperature. Spectra were recorded at intervals during this process. X-ray Absorption Near-edge Structure (XANES) measurements were made in transmission mode at the Fe K, Ni K and Co K-edges, using the order sorting double crystal Si (111) monochromator (4≤hν≤13 keV) on station 7.1. XPS data were collected using the Scienta ESCA 300 system at the RUSTI facility (CLRC Daresbury Laboratory), which incorporates a monochromated Al K α source.
Discussion
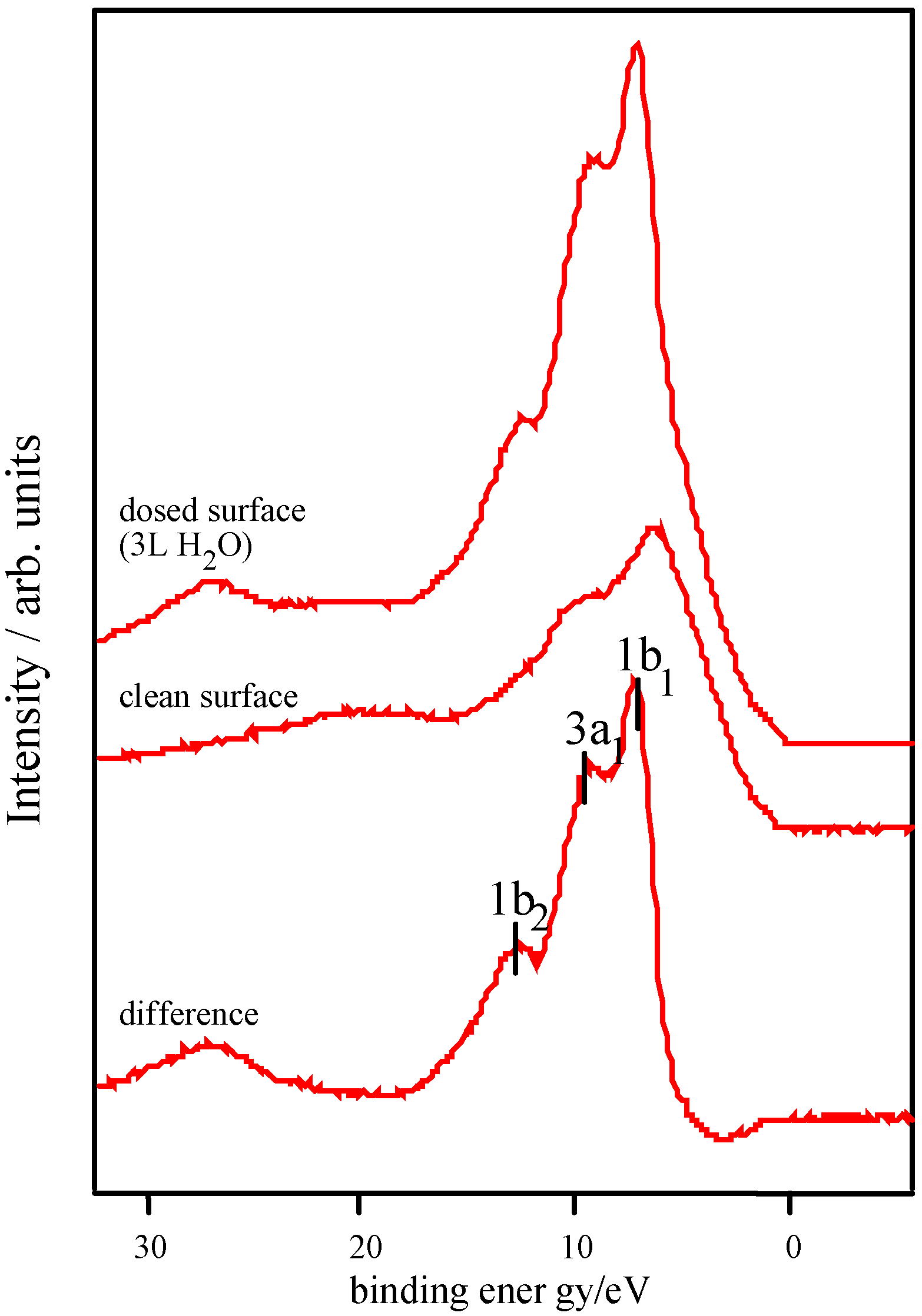
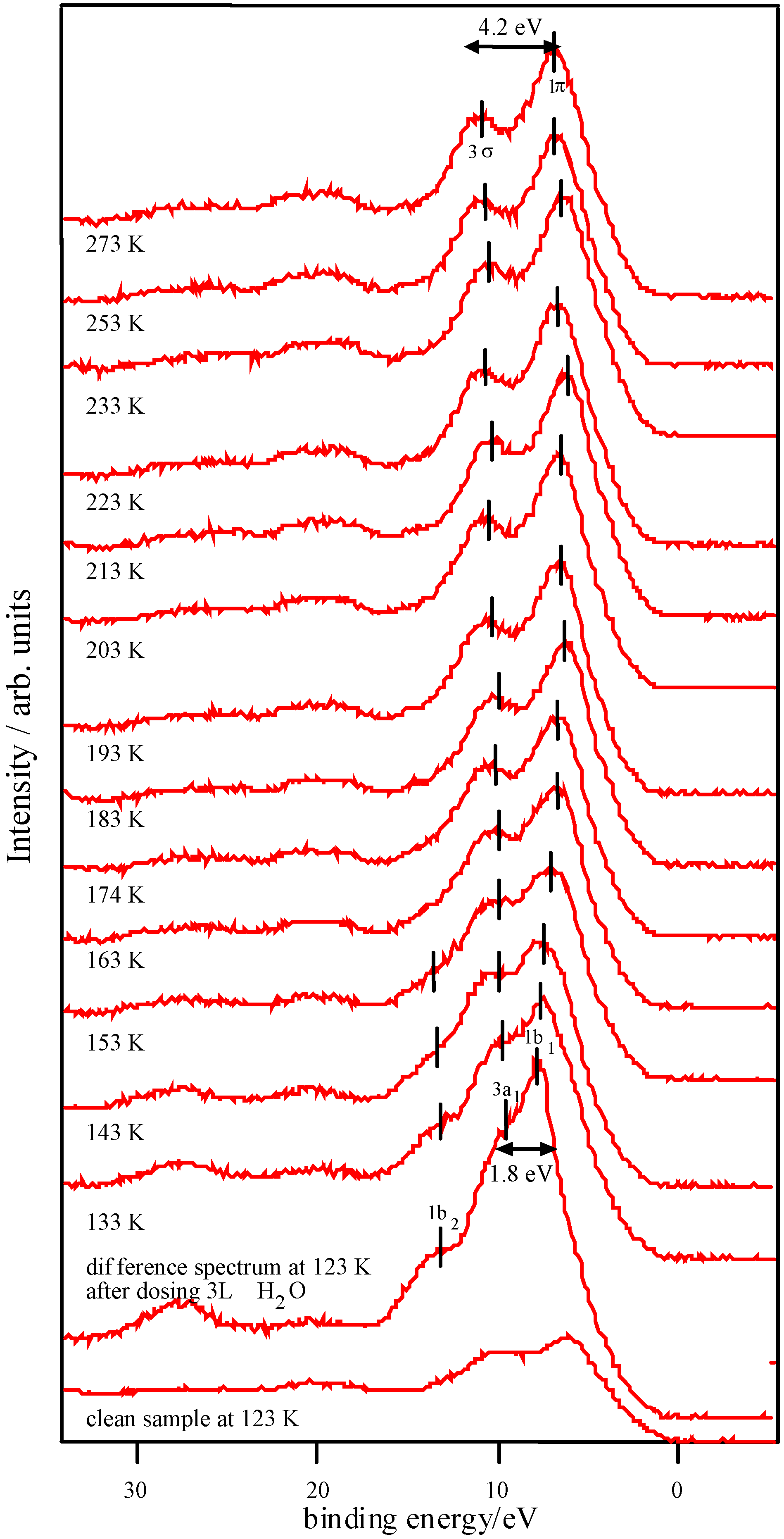
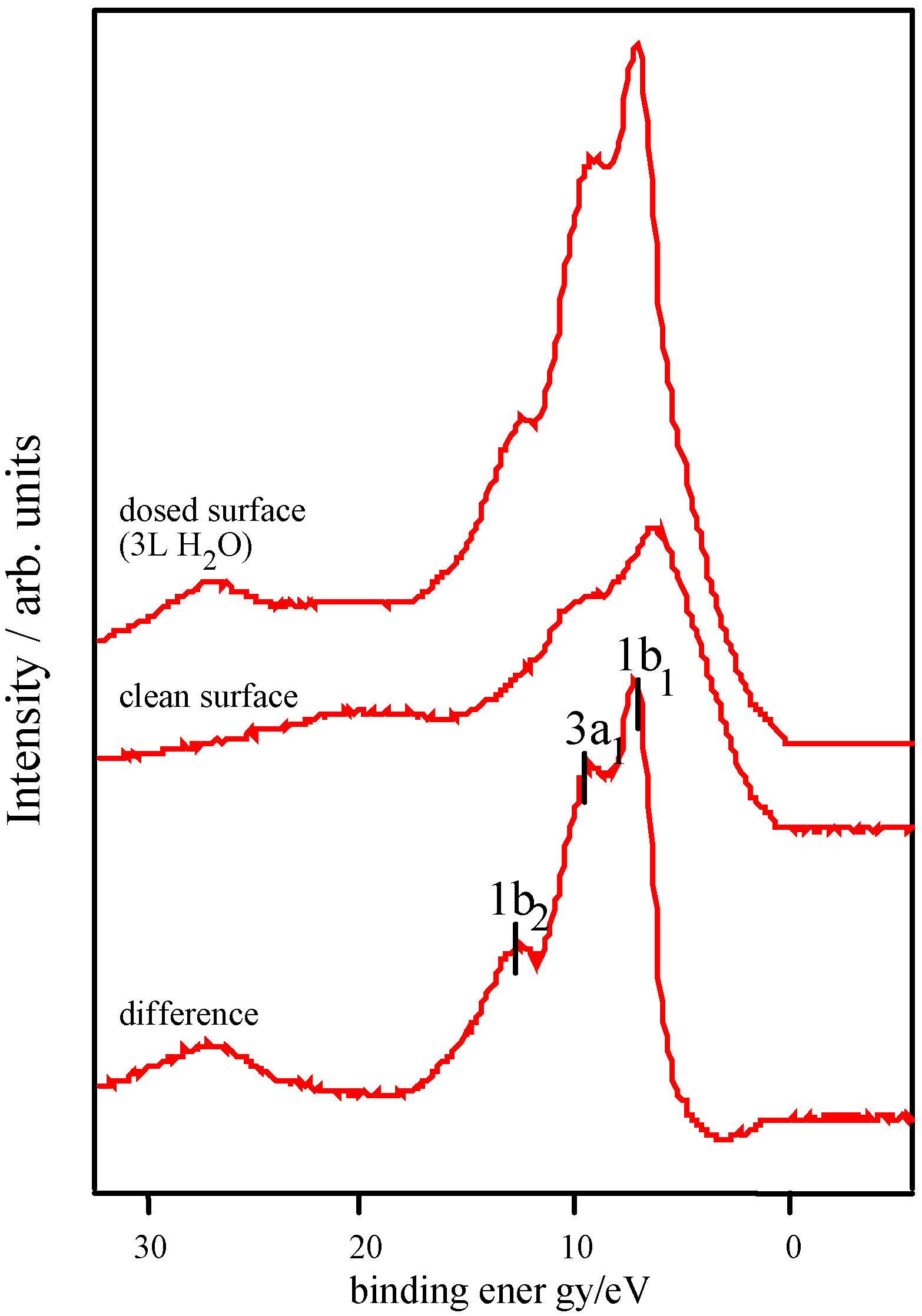
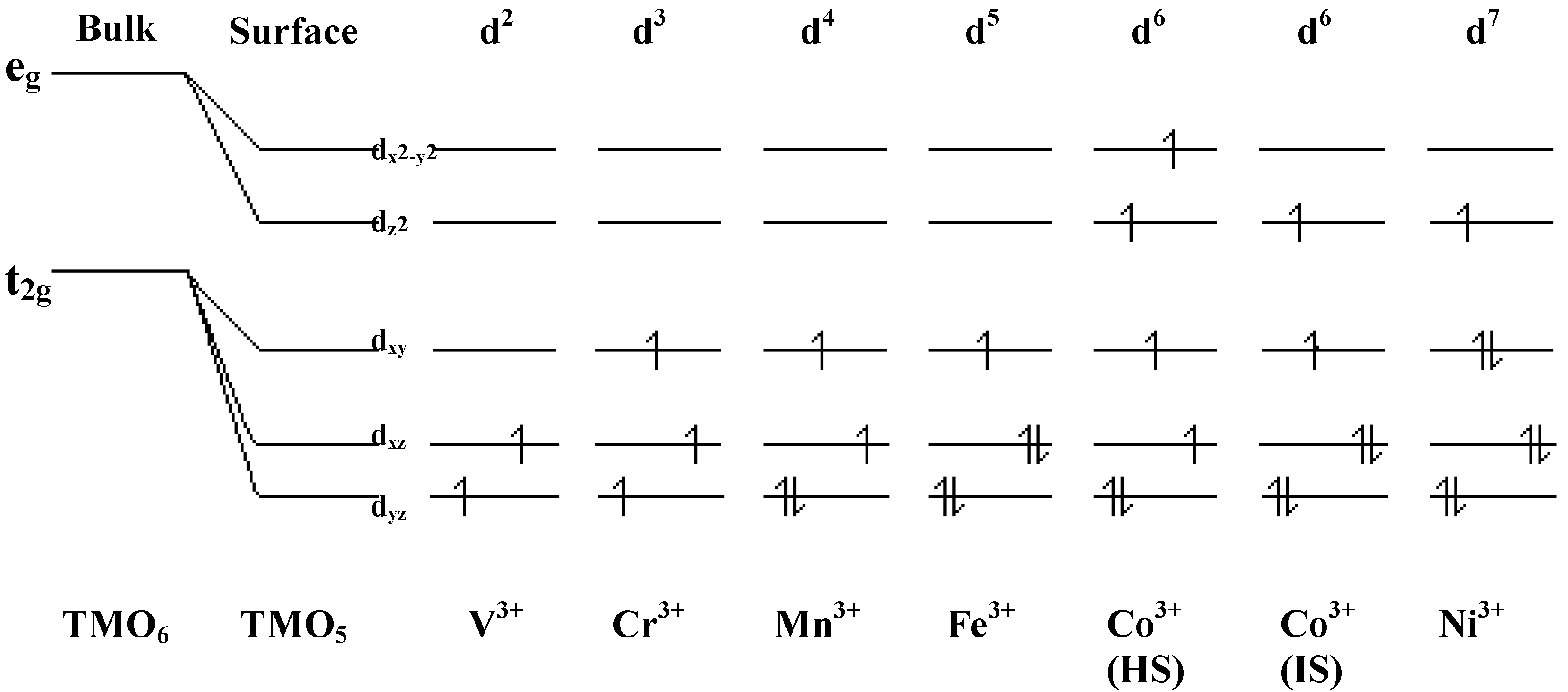
Adsorption studies indicate that the ability of these surfaces to chemisorb water changes with transition metal content. This may be associated with the changing d-electron count of the cations as Co is replaced by Ni or Fe. In cases of strong chemisorption, the interaction between water and transition metal cations on oxide surfaces is of a donor-acceptor type, typically with direct donation from the filled 3a
1 orbital of water into any empty metal 3d levels of the correct symmetry (for an octahedrally coordinated ion, dz
2, e
g). This leads to a bonding stabilisation of the donor levels, and an antibonding destabilisation of the acceptor metal d level [
10]. In the Co containing samples, Co
3+ (d
6) exists in either a high spin (HS) or (predominantly) in an intermediate spin (IS) state [
6], with only partial occupancy of the e
g levels (which are further depopulated on Sr doping, forming some Co
4+). In the Fe-containing samples, the d electron count is lowered, and XANES indicates that the Fe valency in the absence of Sr is Fe
3+ (d
5), with some Fe
4+ (d
4) formed on Sr-doping. In contrast, for Ni containing samples, EXAFS indicates a bondlength intermediate between that of Ni
2+-O and Ni
3+-O, i.e. a raised d electron count of between d
8 and d
7 [
9], and XANES indicates that this does not change on Sr-doping. We may expect that at the surface where adsorption occurs, the regular octahedral coordination of the transition metal ions is broken by the surface, and indeed, cluster calculations by Kojima
et al. [
11] for LaCoO
3 and LaFeO
3 indicate that stable clusters of the type (TMO
5)
7- exist. This raises the degeneracy of both the t
2g and the e
g levels. Of particular interest here is the prediction that Fe
3+ (d
5) exists at the surface not as HS but in a low spin (LS) state with an unoccupied d
z2 orbital [
12], which would lead to a large donor-acceptor interaction on binding water. This is illustrated in
Figure 8 [
12]. In contrast Ni
2+ (LS) has a filled d
z2 level, and thus water might be expected to be essentially physisorbed, as is observed. Overall, we would therefore expect water to bind less strongly to Ni-containing samples than to those containing Fe, and this appears to be borne out by the data contained in
Table 1. We have noted in earlier work that replacing Co by Cu in these systems also results in a decrease in the surface reactivity with water [
13].
Figure 8.
d electron configurations for surface TM3+ ions, adapted from reference 12.
Figure 8.
d electron configurations for surface TM3+ ions, adapted from reference 12.
The catalysis results indicate that the Fe-containing materials decompose peroxide at a substantially higher rate than those containing Ni/Co or Co. The strong reaction between water and Fe-containing samples may aid the leaching of Fe into solution during the reaction, which as the reaction medium is strongly acidic, would exist in solution as Fe
2+ [
14]. Iron in a divalent state is known to readily decompose peroxide under acidic conditions via a Fenton type reaction [
15,
16]:
XPS and AA give clear evidence for leaching of transition metals into solution during reaction. Thus, the increased reactivity of Fe-containing catalysts to water may promote homogeneous decomposition of the oxidant, hydrogen peroxide, and lead to a decrease in activity in epoxidation, as seen in
Table 2.
We may expect any heterogeneous mechanisms for the decomposition of peroxide to be influenced by the bulk defect chemistry, and here XANES indicates that there are marked differences between Fe-doped and Ni-doped samples. Hole doping Fe-containing materials leads to production of Fe
4+. In contrast, in LaCo
1-yNi
yO
3, EXAFS and XANES indicate that Ni is present as mixed Ni
2+ and Ni
3+ [
9]. On hole-doping with Sr, the introduced holes appear to be compensated by the creation of oxygen vacancies [
9] and some Ni
2+ remains. These results are consistent with those of Rajeev
et al. for this phase [
17]. TPD studies of these perovskites have indicated the presence of two types of oxygen, α-oxygen and β-oxygen [
18]. The former desorbs at low temperatures (< 300°C) and is associated with loosely-bound absorbed oxygen accommodated in oxygen vacancy sites in the lattice [
18], and implicated in catalytic activity. Studies of La
1-xSr
xCo
1-yFe
yO
3 [
19,
20] have shown that the amount of α-oxygen increases on Sr-doping, stabilizing the Fe
4+ created on hole doping. In contrast, the amount of α-oxygen in Ni-doped catalysts is relatively low [
20], consistent with the XANES measurements. A large supply of such oxygen is important to heterogeneous peroxide decomposition, thought to occur via a two-step mechanism proposed by Zhang [
19]:
Heterogeneous peroxide decomposition is therefore favoured in Fe-containing samples. Thus, both homogeneous and heterogeneous pathways to the unwanted peroxide decomposition reaction are favoured over Fe-containing samples. In contrast, we expect the desired epoxidation reaction to be favoured in the Ni- and Co-containing materials. In general, epoxidation by late transition metals is via the production of a nucleophilic epoxide species, HO
2-, and attack of this species on the double bond of the organic starting compound [
21] bound to the surface. While our studies give us no information about the binding site of the crotyl alcohol on the surface, the work of Voorhoeve has shown that oxidation over perovskites of the type studied here occurs most efficiently at transition metal ions having quite full t
2g levels and one or no e
g electrons [
22]. This maximises any synergic bonding between the TM site and unsaturated bonds in the organic starting compound. The most suitable configurations in our case are likely to be Ni
3+ and IS Co
3+ (
Figure 8). Thus catalysts containing Co and Co/Ni are efficient epoxidation catalysts, and are also relatively inefficient in unwanted peroxide decomposition.
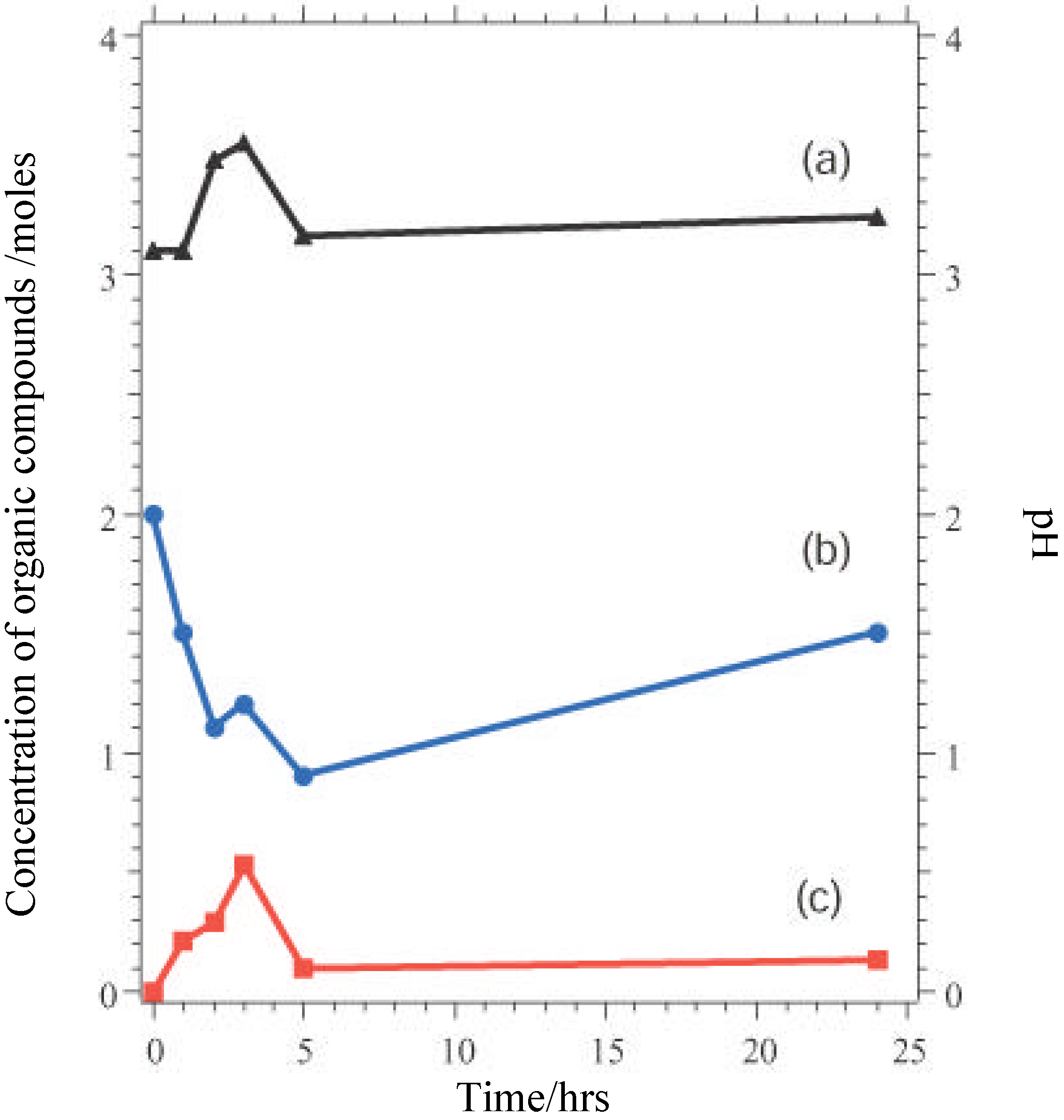
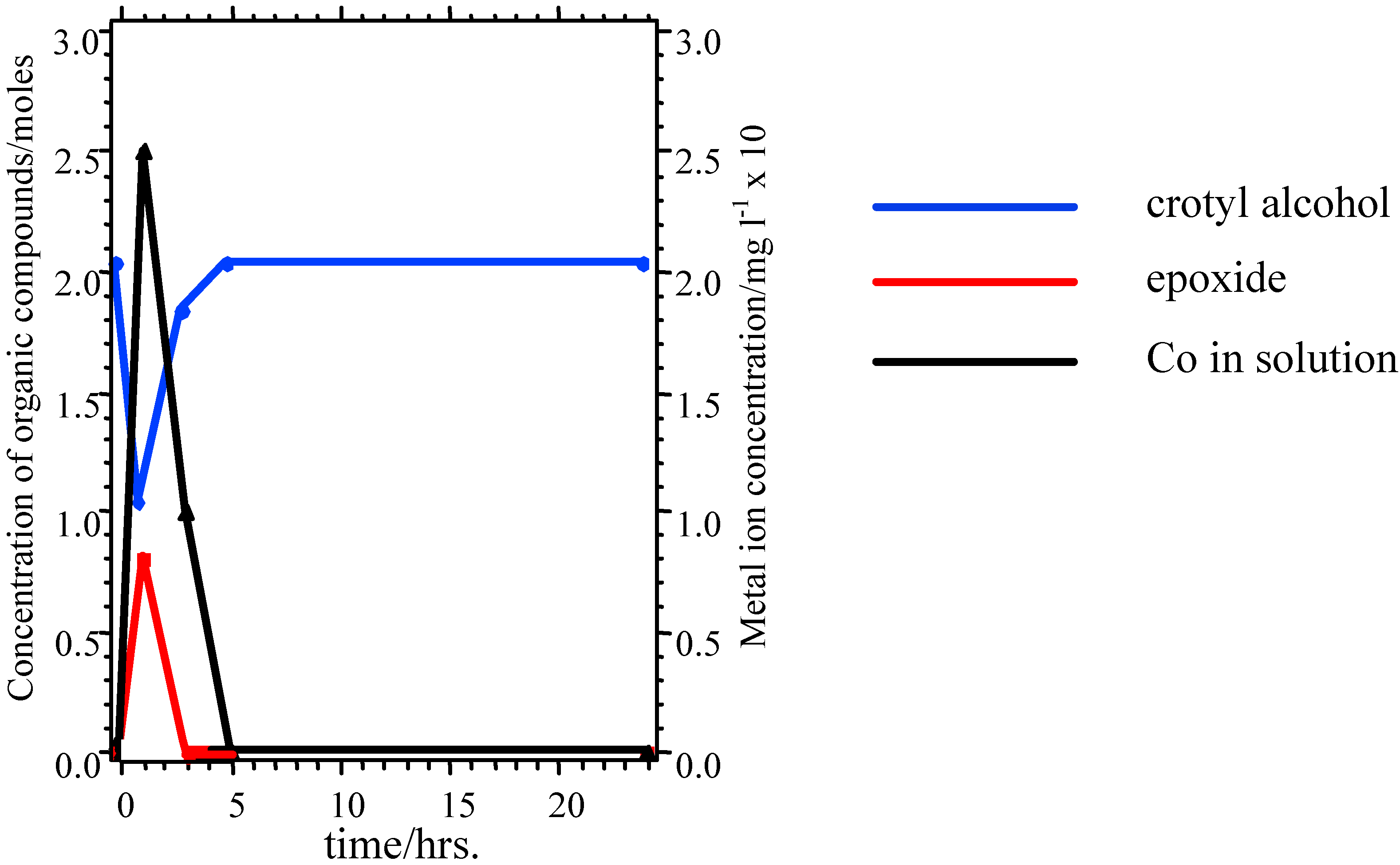
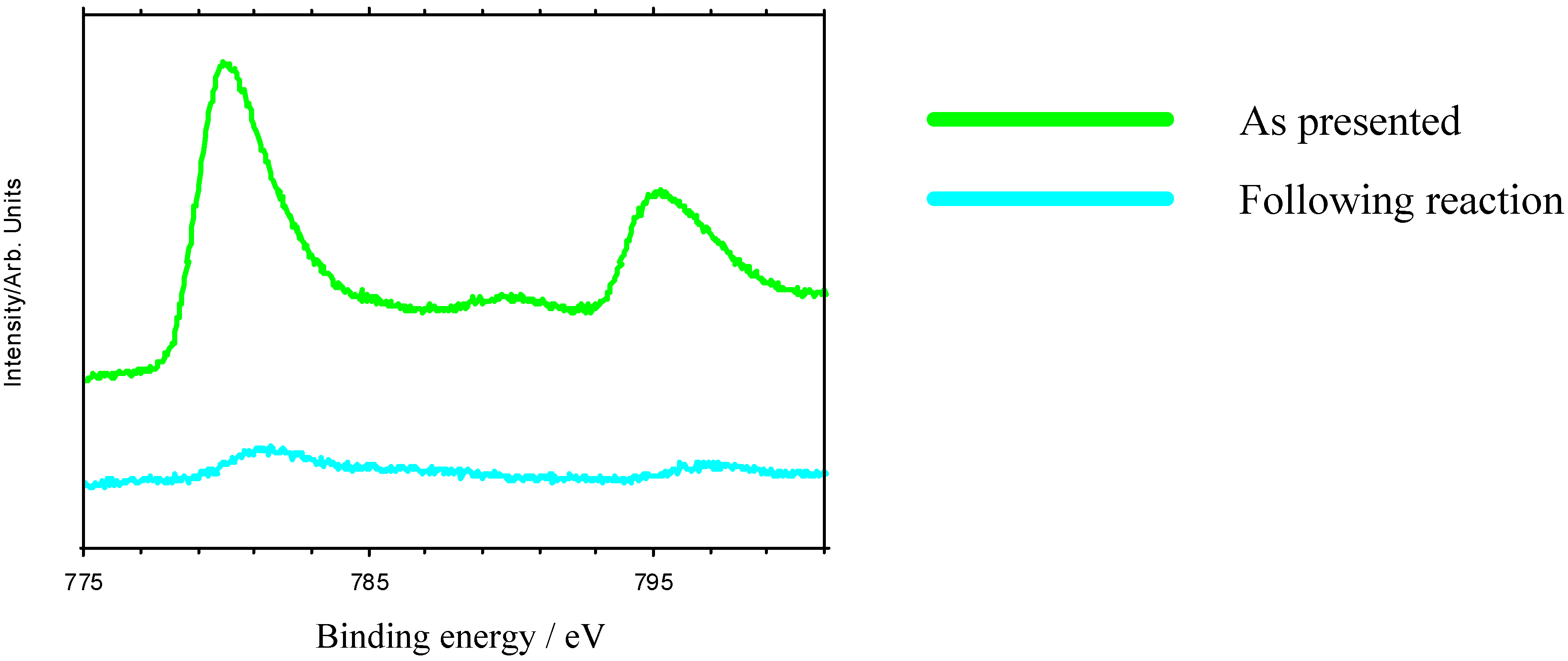
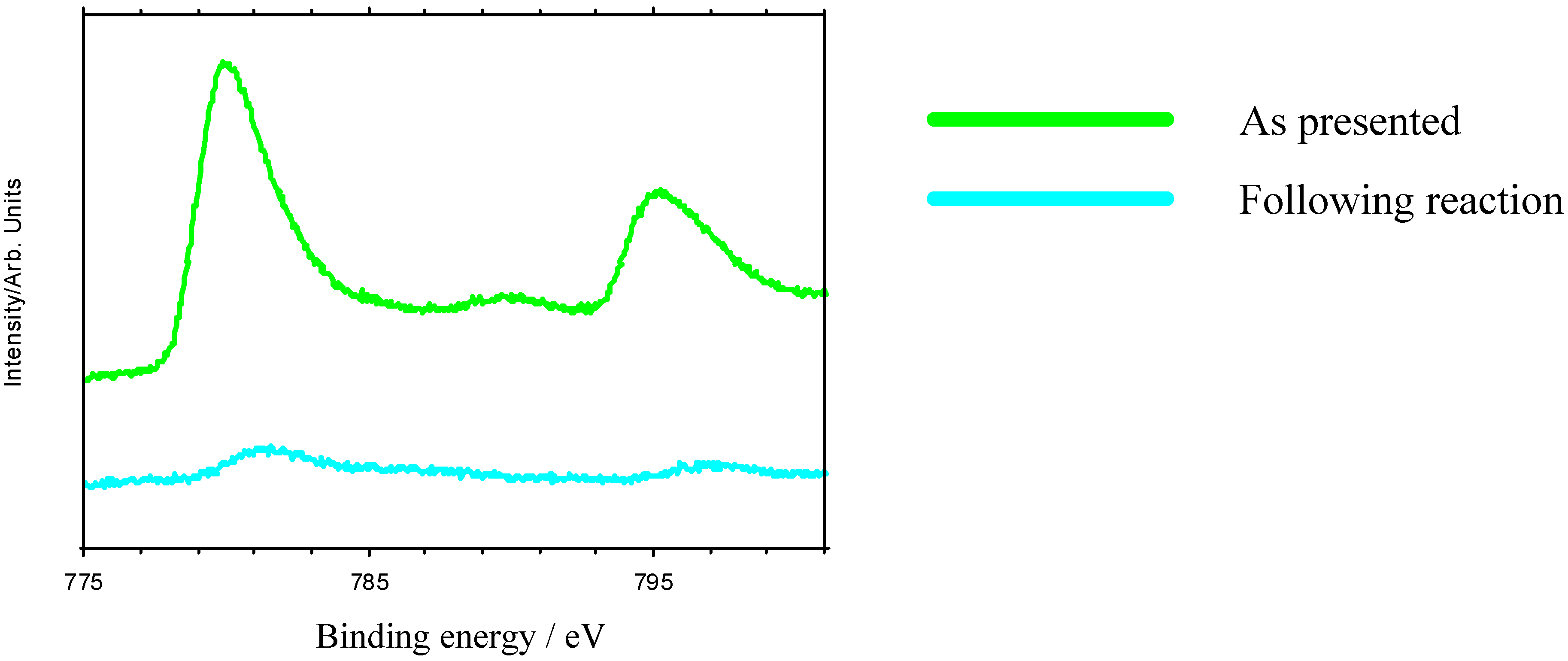
Nevertheless, these materials do show properties which are ultimately undesirable in a working catalyst. GC measurements indicate that there is an initial induction period before reaction begins, while XPS shows that the as-presented catalyst surfaces are initially rich in La-containing contaminant phases. The dissolution of these phases in the highly acidic reaction medium, producing La
3+ in solution may be one factor leading to the increase in pH seen in the initial reaction stages (
Figure 5). The induction period may be associated with the stripping of these phases from the surface to expose the active TM sites. When reaction does commence, marked leaching of the TM ions into solution is observed (
Figure 6), which is undesirable in a working material, and leads to a reduction in the rate of epoxide production typically 3-5 hours into the catalytic reaction. XPS data taken following reaction reveal a marked reduction in surface TM. Reuse tests on these materials show that initially active catalysts retain their activity on reuse, but often with a lengthened induction period before epoxide production begins. This may be because the surface layers of the catalyst, previously depleted in TM, must be stripped before TM-rich layers of the catalyst are again exposed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}