Neuroimmune Semaphorin 4A in Cancer Angiogenesis and Inflammation: A Promoter or a Suppressor?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
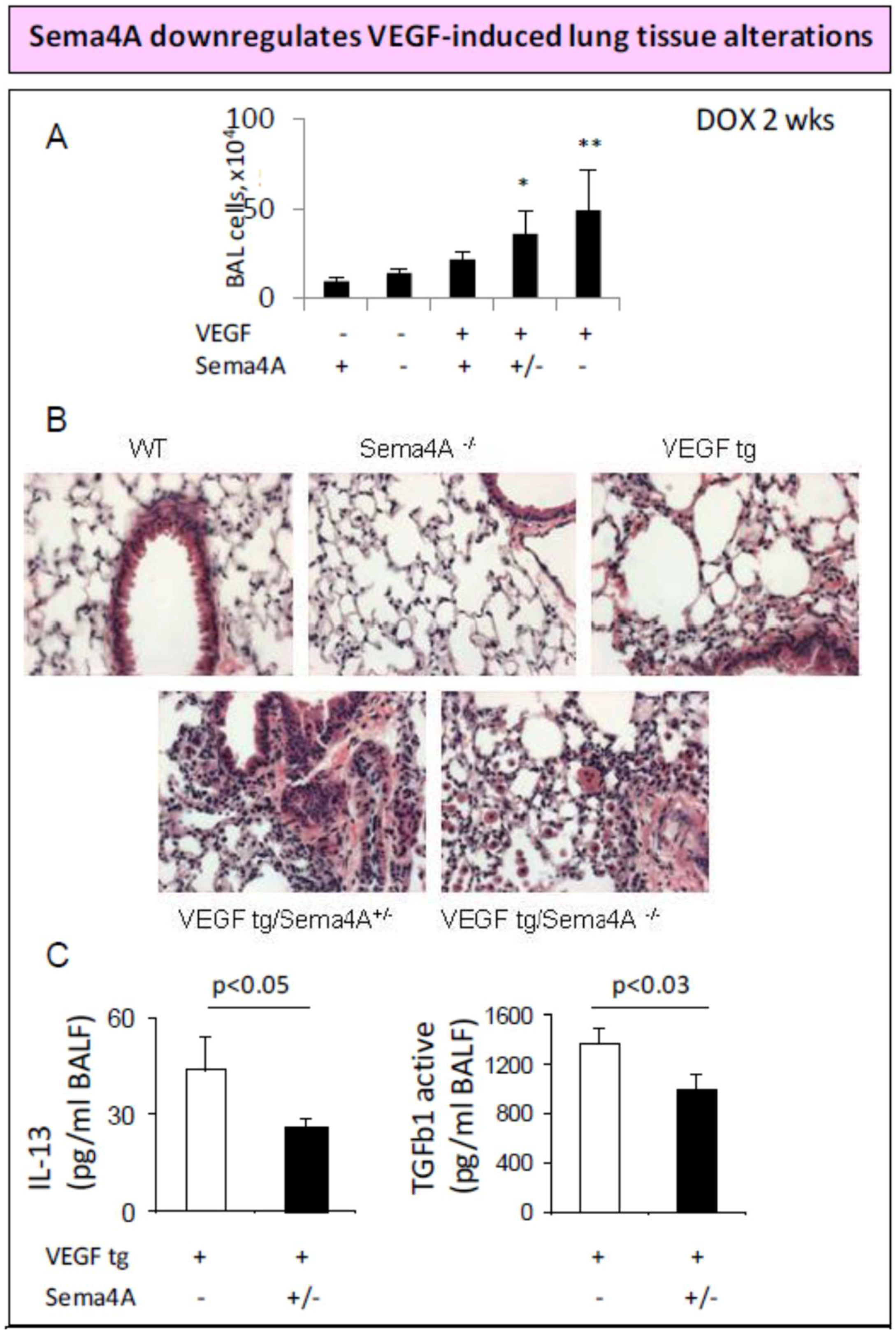
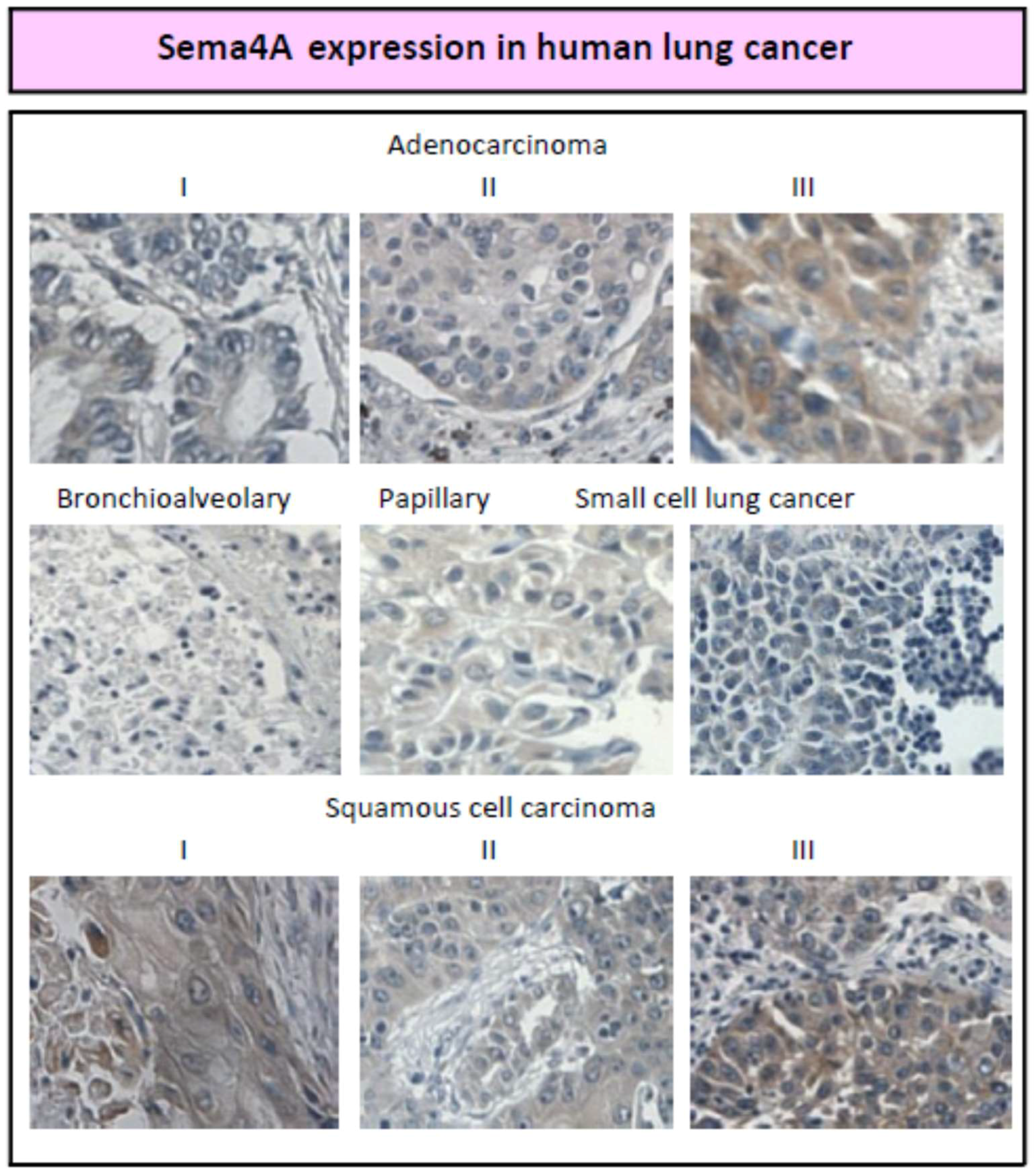
2. Sema4A and Anti-Angiogenic Therapy in Cancer
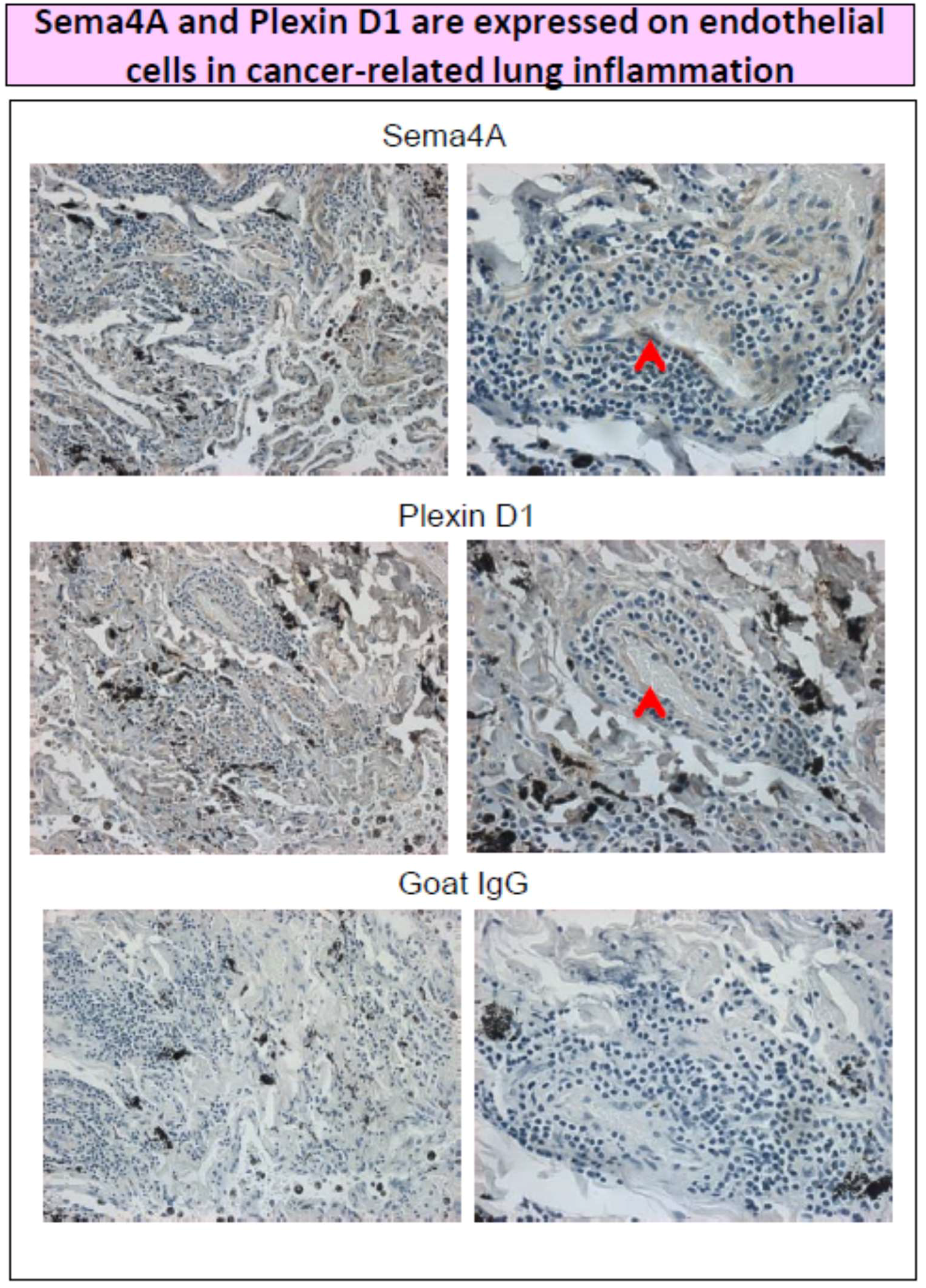
3. Sema4A and Anti-Inflammatory Therapy in Cancer
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Viallard:, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Axitinib: A review in advanced renal cell carcinoma. Drugs 2015, 75, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Sato, A.; Hanaoka, D.; Aonuma, K. Acute aortic dissection with sporadic aortic calcifications during chemotherapy with sunitinib. J. Vasc. Surg. Cases Innov. Tech. 2018, 4, 147. [Google Scholar] [CrossRef] [PubMed]
- Cereda, V.; Formica, V.; Roselli, M. Issues and promises of bevacizumab in prostate cancer treatment. Expert Opin. Biol. Ther. 2018, 18, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Sproat, C.; Kwok, J.; Tanna, N. Axitinib-related osteonecrosis of the jaw. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2017, 124, e257–e260. [Google Scholar] [CrossRef]
- Smith, E.P.; Shanks, K.; Lipsky, M.M.; DeTolla, L.J.; Keegan, A.D.; Chapoval, S.P. Expression of neuroimmune semaphorins 4A and 4D and their receptors in the lung is enhanced by allergen and vascular endothelial growth factor. BMC Immunol. 2011, 12, 30. [Google Scholar] [CrossRef]
- Lee, C.G.; Link, H.; Baluk, P.; Homer, R.J.; Chapoval, S.; Bhandari, V.; Kang, M.J.; Cohn, L.; Kim, Y.K.; McDonald, D.M.; et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat. Med. 2004, 10, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Kumanogoh, A.; Kikutani, H. Immune semaphorins: A new area of semaphorin research. J. Cell Sci. 2003, 116, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kumanogoh, A.; Kikutani, H. Semaphorins and their receptors in immune cell interactions. Nat. Immunol. 2008, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kumanogoh, A.; Shikina, T.; Suzuki, K.; Uematsu, S.; Yukawa, K.; Kashiwamura, S.; Tsutsui, H.; Yamamoto, M.; Takamatsu, H.; Ko-Mitamura, E.P.; et al. Nonredundant roles of Sema4A in the immune system: Defective T cell priming and Th1/Th2 regulation in Sema4A-deficient mice. Immunity 2005, 22, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, S.; Sabatos, C.A.; Xiao, S.; Illes, Z.; Cha, E.K.; Sobel, R.A.; Zheng, X.X.; Strom, T.B. Kuchroo VK: Tim-2 regulates T helper type 2 responses and autoimmunity. J. Exp. Med. 2005, 202, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Woo, S.R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, K.; Tanaka, T.; Yoshida, K.; Takeuchi, N.; Ito, T.; Takamatsu, H.; Kikutani, H.; Kumanogoh, A. Sema4A induces cell morphological changes through B-type plexin-mediated signaling. Int. J. Mol. Med. 2010, 25, 225–230. [Google Scholar] [CrossRef]
- Toyofuku, T.; Yabuki, M.; Kamei, J.; Kamei, M.; Makino, N.; Kumanogoh, A.; Hori, M. Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J. 2007, 26, 1373–1384. [Google Scholar] [CrossRef]
- Lu, N.; Li, Y.; Zhang, Z.; Xing, J.; Sun, Y.; Yao, S.; Chen, L. Human Semaphorin-4A drives Th2 responses by binding to receptor ILT-4. Nat. Commun. 2018, 9, 742. [Google Scholar] [CrossRef]
- Holl, E.K.; Roney, K.E.; Allen, I.C.; Steinbach, E.; Arthur, J.C.; Buntzman, A.; Plevy, S.; Frelinger, J.; Ting, J.P. Plexin-B2 and Plexin-D1 in dendritic cells: Expression and IL-12/IL-23p40 production. PLoS ONE 2012, 7, e43333. [Google Scholar] [CrossRef]
- Ito, D.; Nojima, S.; Nishide, M.; Okuno, T.; Takamatsu, H.; Kang, S.; Kimura, T.; Yoshida, Y.; Morimoto, K.; Maeda, Y.; et al. mTOR Complex Signaling through the SEMA4A-Plexin B2 Axis Is Required for Optimal Activation and differentiation of CD8+ T Cells. J. Immunol. 2015, 195, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Nkyimbeng-Takwi, E.H.; Shanks, K.; Smith, E.; Iyer, A.; Lipsky, M.M.; Detolla, L.J.; Kikutani, H.; Keegan, A.D.; Chapoval, S.P. Neuroimmune semaphorin 4A downregulates the severity of allergic response. Mucosal. Immunol. 2012, 5, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Morihana, T.; Goya, S.; Mizui, M.; Yasui, T.; Prasad, D.V.; Kumanogoh, A.; Tamura, M.; Shikina, T.; Maeda, Y.; Iwamoto, Y.; et al. An inhibitory role for Sema4A in antigen-specific allergic asthma. J. Clin. Immunol. 2013, 33, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P.; Werder, R.B.; Loh, Z.; Sikder, M.A.A.; Curren, B.; Zhang, V.; Rogers, M.J.; Lane, K.; Simpson, J.; Mazzone, S.B.; Spann, K.; et al. Plasmacytoid dendritic cells protect from viral bronchiolitis and asthma through semaphorin 4a-mediated T reg expansion. J. Exp. Med. 2018, 215, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Koda, T.; Okuno, T.; Takata, K.; Honorat, J.A.; Kinoshita, M.; Tada, S.; Moriya, M.; Sakoda, S.; Mochizuki, H.; Kumanogoh, A.; et al. Sema4A inhibits the therapeutic effect of IFN-β in EAE. J. Neuroimmunol. 2014, 268, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Makino, N.; Toyofuku, T.; Takegahara, N.; Takamatsu, H.; Okuno, T.; Nakagawa, Y.; Kang, S.; Nojima, S.; Hori, M.; Kikutani, H.; et al. Involvement of Sema4A in the progression of experimental autoimmune myocarditis. FEBS Lett. 2008, 582, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Chapoval, S.P. Semaphorin 4A as novel regulator and promising therapeutic target in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 313. [Google Scholar] [CrossRef]
- Wang, L.; Song, G.; Zheng, Y.; Tan, W.; Pan, J.; Zhao, Y.; Chang, X. Expression of Semaphorin 4A and its potential role in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 227. [Google Scholar] [CrossRef]
- Rice, D.S.; Huang, W.; Jones, H.A.; Hansen, G.; Ye, G.L.; Xu, N.; Wilson, E.A.; Troughton, K.; Vaddi, K.; Newton, R.C.; et al. Severe retinal degeneration associated with disruption of semaphorin 4A. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2767–2777. [Google Scholar] [CrossRef]
- Yukawa, K.; Tanaka, T.; Bai, T.; Ueyama, T.; Owada-Makabe, K.; Tsubota, Y.; Maeda, M.; Suzuki, K.; Kikutani, H.; Kumanogoh, A. Semaphorin 4A induces growth cone collapse of hippocampal neurons in a Rho/Rho-kinase-dependent manner. Int. J. Mol. Med. 2005, 16, 115–118. [Google Scholar] [CrossRef]
- Meda, C.; Molla, F.; De Pizzol, M.; Regano, D.; Maione, F.; Capano, S.; Locati, M.; Mantovani, A.; Latini, R.; Bussolino, F.; et al. Semaphorin 4A exerts a proangiogenic effect by enhancing vascular endothelial growth factor-A expression in macrophages. J. Immunol. 2012, 188, 4081–4092. [Google Scholar] [CrossRef] [PubMed]
- Leitner, D.F.; Stoute, J.A.; Landmesser, M.; Neely, E.; Connor, J.R. The HFE genotype and a formulated diet controlling for iron status attenuate experimental cerebral malaria in mice. Int. J. Parasitol. 2015, 45, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Klampfl, P.; Holzapfel, S.; Janecke, A.R.; Ulz, P.; Renner, W.; Kashofer, K.; Nojima, S.; Leitner, A.; Zebisch, A.; et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat. Commun. 2014, 5, 5191. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Pecqueux, M.; Halama, N.; Dienemann, H.; Muley, T.; Pfannschmidt, J.; Lasitschka, F.; Klupp, F.; Schmidt, T.; Rahbari, N.; et al. Tumour-site-dependent expression profile of angiogenic factors in tumour-associated stroma of primary colorectal cancer and metastases. Br. J. Cancer 2014, 110, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. Hostile takeover: How tumours hijack pre-existing vascular environments to thrive. J. Pathol. 2017, 242, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Gardner, V.; Madu, C.O.; Lu, Y. Anti-VEGF Therapy in Cancer: A Double-Edged Sword. In Physiologic and Pathologic Angiogenesis. Signaling Mechanisms and Targeted Therapy; InTechOpen: London, UK, 2017; pp. 385–410. [Google Scholar]
- Gressett, S.M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Kumanogoh, A. The role of Sema4A in angiogenesis, immune responses, carcinogenesis, and retinal systems. Cell Adh. Migr. 2016, 10, 692–699. [Google Scholar] [CrossRef]
- Nkyimbeng-Takwi, E.; Chapoval, S.P. Biology and function of neuroimmune semaphorins 4A and 4D. Immunol. Res. 2011, 50, 10–21. [Google Scholar] [CrossRef]
- Neufeld, G.; Mumblat, Y.; Smolkin, T.; Toledano, S.; Nir-Zvi, I.; Ziv, K.; Kessler, O. The role of the semaphorins in cancer. Cell Adh. Migr. 2016, 10, 652–674. [Google Scholar] [CrossRef]
- Basile, J.R.; Afkhami, T.; Gutkind, J.S. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol. Cell Biol. 2005, 25, 6889–6898. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, Z.; Zhang, Z.; Li, J.Y.; Cui, J.; Shi, W.P.; Dong, X.W.; Yuan, L.; Lin, P.; Chen, Z.N.; et al. Aberrant Expression of miR-362 Promotes Lung Cancer Metastasis through Downregulation of Sema3A. J. Immunol. Res. 2018, 2018, 1687097. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.F.; Ye, L.; Jiang, W.G. The Plexin-B family and its role in cancer progression. Histol. Histopathol. 2014, 29, 151–165. [Google Scholar] [PubMed]
- Maione, F.; Molla, F.; Meda, C.; Latini, R.; Zentilin, L.; Giacca, M.; Seano, G.; Serini, G.; Bussolino, F.; Giraudo, E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J. Clin. Investig. 2009, 119, 3356–3372. [Google Scholar] [CrossRef] [PubMed]
- Kutschera, S.; Weber, H.; Weick, A.; De Smet, F.; Genove, G.; Takemoto, M.; Prahst, C.; Riedel, M.; Mikelis, C.; Baulande, S.; et al. Differential endothelial transcriptomics identifies semaphorin 3G as a vascular class 3 semaphorin. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Finisguerra, V.; Capparuccia, L.; Camperi, A.; Swiercz, J.M.; Rizzolio, S.; Rolny, C.; Christensen, C.; Bertotti, A.; Sarotto, I.; et al. Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J. Clin. Investig. 2010, 120, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Lanahan., A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; Toomre, D.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Drabkin, H.A. The role of semaphorins in lung cancer. Clin. Lung Cancer. 2001, 3, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Loginov, V.I.; Dmitriev, A.A.; Senchenko, V.N.; Pronina, I.V.; Khodyrev, D.S.; Kudryavtseva, A.V.; Krasnov, G.S.; Gerashchenko, G.V.; Chashchina, L.I.; Kazubskaya, T.P.; et al. Tumor Suppressor Function of the SEMA3B Gene in Human Lung and Renal Cancers. PLoS ONE 2015, 10, e0123369. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Yin, K.; Ren, G.; Zhang, Y. The crucial role of SEMA3F in suppressing the progression of oral squamous cell carcinoma. Cell Mol. Biol. Lett. 2017, 22, 32. [Google Scholar] [CrossRef]
- Ghelfi, E.; Yu, C.W.; Elmasri, H.; Terwelp, M.; Lee, C.G.; Bhandari, V.; Comhair, S.A.; Erzurum, S.C.; Hotamisligil, G.S.; Elias, J.A.; et al. Fatty acid binding protein 4 regulates VEGF-induced airway angiogenesis and inflammation in a transgenic mouse model: Implications for asthma. Am. J. Pathol. 2013, 182, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Flannery, E.; Duman-Scheel, M. Semaphorins at the interface of development and cancer. Curr. Drug Targets 2009, 10, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Tamagnone, L. Semaphorins in cancer: Biological mechanisms and therapeutic approaches. Semin. Cell Dev. Biol. 2013, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Gioelli, N.; Maione, F.; Camillo, C.; Ghitti, M.; Valdembri, D.; Morello, N.; Darche, M.; Zentilin, L.; Cagnoni, G.; Qiu, Y.; et al. A rationally designed NRP1-independent superagonist SEMA3A mutant is an effective anticancer agent. Sci. Transl. Med. 2018, 10, eaah4807. [Google Scholar] [CrossRef] [PubMed]
- Wallerius, M.; Wallmann, T.; Bartish, M.; Östling, J.; Mezheyeuski, A.; Tobin, N.P.; Nygren, E.; Pangigadde, P.; Pellegrini, P.; Squadrito, M.L.; et al. Guidance Molecule SEMA3A Restricts Tumor Growth by Differentially Regulating the Proliferation of Tumor-Associated Macrophages. Cancer Res. 2016, 76, 3166–3178. [Google Scholar] [CrossRef]
- Lavi, N.; Kessler, O.; Ziv, K.; Nir-Zvi, I.; Mumblat, Y.; Eiza, N.; Paran, Y.; Brenner, B.; Vadasz, Z.; Neufeld, G. Semaphorin-3A inhibits multiple myeloma progression in a mouse model. Carcinogenesis 2018, 39, 1283–1291. [Google Scholar] [CrossRef]
- Luchino, J.; Hocine, M.; Amoureux, M.C.; Gibert, B.; Bernet, A.; Royet, A.; Treilleux, I.; Lécine, P.; Borg, J.P.; Mehlen, P.; et al. Semaphorin 3E suppresses tumor cell death triggered by the plexin D1 dependence receptor in metastatic breast cancers. Cancer Cell. 2013, 24, 673–685. [Google Scholar] [CrossRef]
- Movassagh, H.; Shan, L.; Duke-Cohan, J.S.; Halayko, A.J.; Uzonna, J.E.; Gounni, A.S. Semaphorin 3E Alleviates Hallmarks of House Dust Mite-Induced Allergic Airway Disease. Am. J. Pathol. 2017, 187, 1566–1576. [Google Scholar] [CrossRef]
- Movassagh, H.; Shan, L.; Mohammed, A.; Halayko, A.J.; Gounni, A.S. Semaphorin 3E Deficiency Exacerbates Airway Inflammation, Hyperresponsiveness, and Remodeling in a Mouse Model of Allergic Asthma. J. Immunol. 2017, 198, 1805–1814. [Google Scholar] [CrossRef]
- Mogie, G.; Shanks, K.; Nkyimbeng-Takwi, E.H.; Smith, E.; Davila, E.; Lipsky, M.M.; DeTolla, L.J.; Keegan, A.D.; Chapoval, S.P. Neuroimmune semaphorin 4A as a drug and drug target for asthma. Int. Immunopharmacol. 2013, 17, 568–575. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Y.; Tian, W.; Wang, F.; Lv, X.; Wang, M.; Sun, T.; Zhang, J.; Wang, L.; Han, M. Sema4A Responds to Hypoxia and Is Involved in Breast Cancer Progression. Biol. Pharm. Bull. 2018, 41, 1791–1796. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, A.S.; Chapoval, S.P. Neuroimmune Semaphorin 4A in Cancer Angiogenesis and Inflammation: A Promoter or a Suppressor? Int. J. Mol. Sci. 2019, 20, 124. https://doi.org/10.3390/ijms20010124
Iyer AS, Chapoval SP. Neuroimmune Semaphorin 4A in Cancer Angiogenesis and Inflammation: A Promoter or a Suppressor? International Journal of Molecular Sciences. 2019; 20(1):124. https://doi.org/10.3390/ijms20010124
Chicago/Turabian StyleIyer, Apoorva S., and Svetlana P. Chapoval. 2019. "Neuroimmune Semaphorin 4A in Cancer Angiogenesis and Inflammation: A Promoter or a Suppressor?" International Journal of Molecular Sciences 20, no. 1: 124. https://doi.org/10.3390/ijms20010124
APA StyleIyer, A. S., & Chapoval, S. P. (2019). Neuroimmune Semaphorin 4A in Cancer Angiogenesis and Inflammation: A Promoter or a Suppressor? International Journal of Molecular Sciences, 20(1), 124. https://doi.org/10.3390/ijms20010124