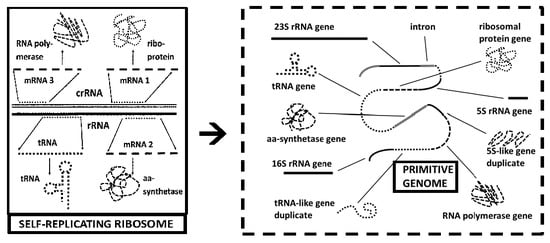
2.1. Eukaryotic and Prokaryotic Genomes Contain Unexpectedly Large Numbers of rRNA-Like Sequences
All cellular genomes contain unexpectedly large numbers of rRNA-like sequences, an observation that was first reported by Mauro and Edelman in 1997 for eukaryotes [
13]: “Many eukaryotic mRNAs contain sequences that resemble segments of 28S and 18S rRNAs, and these rRNA-like sequences are present in both the sense and antisense orientations. Some are similar to highly conserved regions of the rRNAs, whereas others have sequence similarities to expansion segments. In particular, four 18S rRNA-like sequences are found in several hundred different genes, and the location of these four sequences within the various genes is not random. One of these rRNA-like sequences is preferentially located within protein coding regions immediately upstream of the termination codon of a number of genes…. We consider the hypotheses that rRNA-like sequences may have spread throughout eukaryotic genomes and that their presence in primary transcripts may differentially affect gene expression.”
Several lines of evidence supported Mauro and Edelman’s conjecture. Mauro and Edelman themselves demonstrated that regions within mRNAs that were complementary to rRNA bound to rRNAs in the ribosome resulting in translational control of gene products. The Gtx homeodomain protein is one well-characterized example [
14,
15,
16]. Several investigators have also demonstrated the presence of rRNA-like sequences in a wide array of poly-adenylated mRNAs. Poly-adenylation is a process in which adenines are added to the end of mRNA during transcription, to aid in the transport of the mRNA to the ribosome, and to identify the RNA as carrying a translatable message. Thus, the presence of rRNA-like sequences in polyadenylated RNAs provides further evidence of a role for rRNA sequences in identifying functional proteins.
Mauro and Edelman, for example, found that [
13]: “Northern blot analysis of poly(A) RNA from different vertebrates (chicken, cattle, rat, mouse, and human) revealed that a large number of discrete RNA molecules hybridize at high stringency to cloned probes prepared from the 28S or 18S rRNA sequences that were found to match those in mRNAs. Inhibition of polymerase II activity, which prevents the synthesis of most mRNAs, abolished most of the hybridization to the rRNA probes.” Kong, et al. [
17] verified that rRNA-like sequences are unusually abundant in poly-adenylated mRNA. Kong, et al. [
17] also identified two different types of such poly-adenylated rRNA-containing transcripts (PART). Type I PART contain sequences of tens to several hundreds of rRNA-associated nucleotides within the transcript itself, while Type II PART contain very long, sometimes almost complete, rRNA sequences with a 5′ cap and a 3′ polyadenylation sequence. PART I include tRNA synthetases, RNA-binding proteins, insulin-like growth factor binding proteins, tumor necrosis factor, homeobox genes, heat shock 86 protein, connexins, glycine methyltransferase, nuclear respiratory factor 1 (NRF 1), and many other classes of proteins that are essential to cell functions. PART II include mitochondrial leucyl-tRNA synthetase, which is 98% identical to a 396 nucleotide stretch of the 28S rRNA; phosphocan short isoform (RPTP-beta gene), which is 98% of 1537 nucleotides of the 28S rRNA; superoxide dismutase 1 (SOD1) upregulated mRNA 1, which is identical to the first 1700 of 1879 nucleotides of 18S rRNA; human humanin (HN1), which is identical to 1553 nucleotides of the 28S rRNA (see also [
18]); human serine/threonine protein kinase Kp78, CTAK75a, which is 99% identical to 905 nucleotides of the 18S rRNA; mouse ETS-related transcription factor ERF (Erf1), which is 95% identical to a 705 nucleotide stretch of the 18S rRNA; mouse phosphacan short isoform (RPTP-beta gene), which is 98% identical to 1573 nucleotides of the 28S rRNA; human succinate dehydrogenase complex, subunit A, which is 99% identical to 643 nucleotides of the 18S rRNA [
17]. Kong, et al., conclude that, “It appears that rRNA segments are dispersed throughout the genome” [
17].
Additional proteins that share significant portions of rRNA sequences have been identified by other investigators. One is the rDNA-encoded mitochondrial protein Tar1p (transcript antisense to ribosomal RNA) in
Saccharomyces cerevisiae [
19,
20], which regulates glucose-related respiration. Coelho et al. argue that the existence of Tar1p may demonstrate, “that rDNA transcription and mitochondrial function are coordinately regulated in eukaryotic cells” [
19]. Lee, et al. [
18] add that Tar1p is likely to be only one of many such mitochondria-derived rRNA transcripts: “Many of the mRNA species identified from the mitochondria are discrete smaller length ones that do not map to the traditional mitochondrial protein-encoding genes. In particular, multiple such mRNAs are observed from the 16S rRNA region, including the site of the humanin ORF…. Using parallel analysis of RNA ends (PARE) to map the transcript cleavage sites, a plethora of expected and unexpected cleavage sites have been discovered for the mitochondria [
21]. The majority of tRNAs and mRNAs have distinct dominant cleavage sites at the 5′ termini, but intragenic cleavage sites are especially abundant in rRNAs. The latter stages of precursor modification include 3′ polyadenylation of mRNAs, with the exception of the mitochondrial ND6 gene, and rRNAs, in agreement with previous reports [
21,
22]. In addition, mitochondrial rRNAs are transported and found in the cytoplasm with significant biological roles in, for instance,
Drosophila germ cell establishment [
23,
24], and 16S rRNA was found to be localized in the nucleus of human spermatogenic cells [
25]. It is possible that the polyadenylated humanin transcript is exported to the cytoplasm where it can be translated.”
Mignone and Pesole [
26] have also reported very large numbers of rRNA-like sequences in the human genome, but in contradistinction to the papers just cited, have argued that the vast majority (especially of the longer, type II PART) are artifacts of homology searching, and that only a small subset of short, type I PART stand up to critical analysis of the data. Their own data analysis, however, argues against their conclusions, at least from a ribosome-first perspective. They note that while “the distribution of low-similarity matches was similar for both real and simulated rRNA sequences,… interestingly,… the long/highly similar matches can be found only with the actual rRNA sequences” ([
26] p. 150). Oddly, then, they discarded all of the long sequence (type II PART) homologies as artifacts on the basis that, “The great majority of rRNA-like tracts found in human mRNAs were not supported by counterparts in genomic or EST [expressed sequence tag] sequences” ([
26] p. 151)—a statement in direct contradiction to the results of Mauro and Edelman [
13] and Kong [
17]. In any event, Mignone’s and Pesole’s requirement that rRNA-like sequences actually be transcribed is not required by the ribosome-first theory, which demands only that rRNA-like sequences be found in unexpectedly high numbers compared to the numbers of genes that are required to encode ribosome production. It is quite possible that organismal genomes evolved from duplicated rRNAs and subsequently transferred protein-encoding functions to more specialized exon regions. We believe that this is, in fact, is exactly what Mignone and Pesole found. Additionally, our own analysis of the occurrence of rRNA-like sequences in the genome, which we provide below, reveals a possible flaw in the method by which Mignone and Pesole analyzed their results.
A third set of evidence also argues for the presence of rRNA-derived sequences in cellular genomes at unusually high rates. Gene sequences that are complementary to rRNA genes also populate the genome in unexpectedly large numbers, perhaps representing the incorporation of transcription products of rRNA “genes” into the genome. Examples include complementarity between ferritin H mRNA and 28S ribosomal RNA [
27] between the avian myeloblastosis oncogene and eukaryotic 28S ribosomal RNA [
28], and murine 18S rRNA with a wide range of mRNAs [
29]. Such complementary sequences are not predicted by the genome-first or metabolism-first theories of cellular evolution, but they follow directly from the ribosome-first theory in which all six reading frames of rRNA are utilized to encode protein sequences.
Other data also take on different interpretations, depending on the theoretical framework from which they are interpreted. Genome-wide sequencing studies have repeatedly run into the problem of “discovering” new genes and new gene families within or across overlapping genome regions that encode rRNA. The ribosome-first theory of cellular evolution explicitly proposes that such overlaps not only should, but they must exist, since the rRNAs must contain the genes encoding for their own ribosomal proteins. Thus, the protein-encoding regions must be contained within, or overlap, the rRNA sequences [
6,
7,
8]. No such prediction follows from the genome-first or the metabolism-first theories of cellular evolution. In a genome-first theory, the ribosome would have evolved after the core genome in order to make possible protein production, in which case protein-encoding regions that overlapped with rRNAs would interfere with pre-existing gene function. Similarly, if the ribosome evolved to optimize pre-existing protein metabolism, then key protein sequences should be the basis for rRNA evolution rather than rRNAs serving as the basis for protein evolution.
In fact, a robust literature clearly demonstrates that rRNAs do overlap both known and possible protein-encoding sequences, and there is no evidence that specific protein-encoding sequences are the origins of rRNAs. For example, metagenomic studies of RNA expression generally yield a predominance of rRNA-like sequences that can approach 90% of the total RNA sequenced [
30,
31,
32,
33,
34,
35]. Tripp, et al. [
36] have provided a good summary of the type of data that are treated as artefactual: “In the course of analyzing 9,522,746 pyrosequencing reads from 23 stations in the Southwestern Pacific and equatorial Atlantic oceans, it came to our attention that misannotations of rRNA as proteins is now so widespread that the rate of false-positive matching of rRNA pyrosequencing reads to the National Center for Biotechnology Information (NCBI) non-redundant protein database approaches 90%. One conserved portion of 23S rRNA was consistently misannotated often enough to prompt curators at Pfam to create a spurious protein family” ([
36], p. 8792). This “problem” of pseudo-proteins being encoded within rRNA sequences, or overlapping them, is so widespread that programs have been written to aid researchers in eliminating them during genome-wide sequencing experiments (e.g., [
30,
31,
32,
33,
34,
35]). Without challenging whether proteins are actively encoded in rRNA-encoding sequences in prokaryotes and archaea, the ribosome-first theory does force us to consider the possibility that such apparent encodings are fossils of the ribosomal origins of the genome. At the same time, the widespread existence of such inclusions and overlaps makes no sense from other genome origins theories. From an evolutionary perspective, we must confront the question of why so many “spurious” rRNA-like sequences exist.
It is also important to point out that the desire to eliminate “false” protein families from genome-wide sequencing studies is based on three key points. One is the assumption that rRNA-like sequences should not appear ubiquitously in the genome. The ribosome-first theory undercuts that assumption. The second point is that the argument for eliminating rRNA-like sequences is based on a lack of evidence that they are proper annotations. Lack of evidence is not evidence against their possible validity. Yet, this is precisely the current reasoning. As Tripp, et al. point out, “rRNA operons in Bacteria and Archaea are not known to contain naturally expressed protein coding regions that also code for rRNA” ([
36] p. 8792). Tripp, et al., therefore consider that, “Annotations of Bacteria and Archaea proteins embedded in rRNA operons and overlapping with rRNA coding regions within those operons have been
rightly [emphasis added] presumed to be misannotations [
37] and should continue to be, until hard evidence to the contrary emerges” ([
36] p. 8792). From a ribosome-first perspective, we would strongly urge a search for such “hard evidence”, since it seems unlikely that dual-use, overlapping protein-rRNA sequences evolved ubiquitously without some function, either past or present. Moreover, there is no rationale other than a ribosome-first origin, for the genome to explain why rRNA-like sequences would be both ubiquitous and meaningless, and that rationale would hold only if functions originally associated with rRNA genes were off-loaded onto other genes during evolution.
In fact, many dual-use genes encoding both proteins and rRNAs are known in eukaryotes [
13,
20,
24,
37,
38,
39]. Recently, Mumtaz and Couso [
40] have used Ribo-Seq techniques to examine ribosome-bound mRNAs, and they discovered large numbers of rRNA-like sequences bound to the ribosomes that were apparently translated, and which had previously been ignored as “non-coding” sequences: “Earlier bioinformatic approaches had shown the presence of hundreds of thousands of putative small ORFs (smORFs) in eukaryotic genomes, but they had been largely ignored, due to their large numbers and the difficulty in determining their translation and function. Ribo-Seq has revealed that hundreds of putative smORFs within previously assumed long non-coding RNAs (lncRNAs) and UTRs of canonical mRNAs are associated with ribosomes, appearing to be translated” [
40].
One must consider an additional hypothesis, which is that dual-use sequences may have been very common during the emergence of cellular life, but that selection pressures have largely separated rRNA-encoding sequences from protein-encoding sequences, so as to avoid regulatory confusion. The evolutionary problem that must be faced, regardless of which perspective is taken then, is to explain the origins of ubiquitous rRNA-protein overlaps, whether they are non-functional fossils or functional overlaps. This problem currently appears to be intractable from anything other than the ribosome-first theory. Indeed, this evolutionary problem exists for the origins of all rRNA-like genes within genomes. To quote Kong, et al. again: “For type II PART, there is a possibility that some of these RNA may derive from rRNA genes or rRNA. The origins of these types of RNA remain to be elucidated” [
17].
2.2. Genomes Contain Unexpectedly Large Numbers of rRNA- and tRNA-Derived Transposon Sequences
According to the ribosome-first theory, not only should rRNA-like sequences be unusually prevalent in all cellular genomes, so should tRNA-like sequences. This prediction follows from the fact that tRNA-like sequences appear in multiple reading frames within rRNAs [
6,
7,
8,
9,
10,
11]. Indeed, recent research has revealed that prokaryotic and eukaryotic genomes contain large numbers of short interspersed repetitive elements (SINE) derived from fragments of rRNAs and tRNAs (SINE1) or 7SL (Alu) (SINE 2), as well as mammalian-wide interspersed repeats (MIRs). These sequences may provide evidence for how rRNA (or tRNA)-derived ribosome-encoded genes multiplied to form cellular genomes.
Alu (named after the
Arthrobacter luteus restriction endonuclease), or SINE2, are abundant transposable elements within genomes. While the genes encoding ribosomal RNA, tRNAs, and ribosomal proteins (rDNA) comprise only 0.01% of the human genome, and exons comprise only 1.5% of the human genome [
41], tRNA-like Alu and SINES make up about 11% to 15% of mammalian genomes, and are largely located in intron regions [
41,
42]. MIRs make up another 1–2% of most organismal genomes [
43].
Investigators have also found increasing numbers of new types of SINE derived from 5S rRNA (SINE3) and 28S rRNA (SINE 28). Longo, et al. [
44] provide a good summary of the new findings: “Complex eukaryotic genomes are riddled with repeated sequences whose derivation does not coincide with phylogenetic history, and thus is often unknown. Among such sequences, the capacity for transcriptional activity, coupled with the adaptive use of reverse transcription, can lead to a diverse group of genomic elements across taxa, otherwise known as selfish elements or mobile elements. Short interspersed nuclear elements (SINEs) are nonautonomous mobile elements found in eukaryotic genomes, typically derived from cellular RNAs such as tRNAs, 7SL, or 5S rRNA. Here, we identify and characterize a previously unknown SINE derived from the 3′-end of the large ribosomal subunit (LSU or 28S rDNA) and transcribed via RNA polymerase III. This new element, SINE28, is represented in low-copy numbers in the human reference genome assembly, wherein we have identified 27 discrete loci” [
44]. Additional sequences derived from the 28S ribosome can be found scattered throughout the human genome in varying copy numbers [
45,
46].
Highly conserved 5S rRNA-derived SINEs (SINE3) have been reported in human and other mammals that Nishihara, et al. [
47] have called AmnSINE1. “AmnSINE1 has a chimeric structure of a 5S rRNA and a tRNA-derived SINE, and is related to five tRNA-derived SINE families that we characterized here in the coelacanth, dogfish shark, hagfish, and amphioxus genomes. All of the newly described SINE families have a common central domain that is also shared by zebrafish SINE3, and we collectively name them the DeuSINE (Deuterostomia SINE) superfamily. Notably, of the approximately 1000 still identifiable copies of AmnSINE1 in the human genome, 105 correspond to loci phylogenetically highly conserved among mammalian orthologs.” This high degree of conservation strongly suggests evolutionary selection for functionality. Other examples of SINE3 have been found in fruit bats [
48], fungi [
49], and zebrafish [
50] among other organisms [
51]. In zebrafish, Kapitonov and Jurka report that, “SINE3s are transcribed from the type 1 internal pol III promoter. Approximately 10,000 copies of SINE3 elements are present in the zebrafish genome; they constitute approximately 0.4% of the genomic DNA” [
50]. The 1000 or so SINE3 found in the human genome constitute an equivalent percentage.
In light of the fact that SINES and MIRs make up 15% to 20% of many genomes, Frenkel, et al. [
52] and Schmitz [
42] have proposed that SINEs may be the drivers of genome evolution. If that hypothesis is correct, then the further possibility must be entertained that the tRNA and rRNA origins of SINEs and MIRs are molecular “fossils” of the mechanism by which the original cellular genomes were formed through self-replicating, or insertion-amplifying rRNA and tRNA sequences. Such a mechanism would be entirely consistent with a ribosome-first origin of cellular genomes, but once again, this is difficult to explain from other theoretical perspectives.
2.3. tRNA and rRNA Control of Non-Ribosomal Cellular Functions
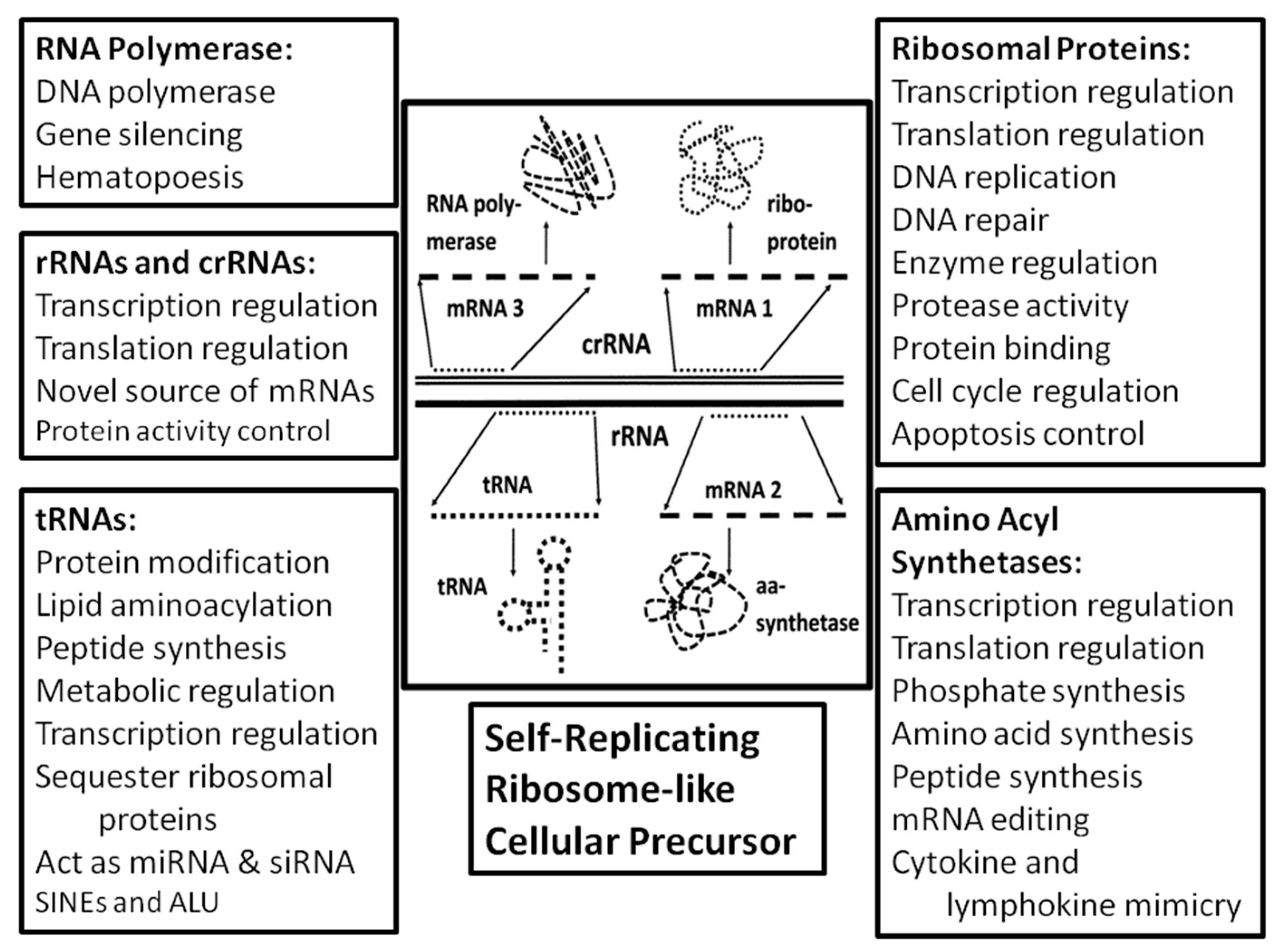
A further unique, testable implication of the ribosome-first theory of genome origins is that ribosomal proteins, rRNAs and tRNAs should be pleiofunctional, playing roles in cellular functions beyond translation. This prediction follows from the assumption that the ribosomal molecules were the foundation from which other core cellular molecules and their functions evolved. The interactivity of the ribosomal complex itself would have assured a high degree of interactivity among the genes and proteins that evolved from it, creating a stable interactome. Therefore ribosomally-derived molecules would have been under significant selection pressure to be retained in primordial cells, and to become adapted to the broader range of functions that the cell required. Evidence for such a scenario is abundant.
Wegrzyn and Wegrzyn [
53] and Katz, et al. [
54] have reviewed studies demonstrating that tRNA has multiple cellular functions beyond is canonical role in protein translation (
Table 1). Various retroplasmids and retroviruses encode a reverse transcriptase that can use a tRNA sequence as the basis for synthesizing a cDNA copies. DNA replication itself is regulated by the accumulation of uncharged tRNAs that bind to, and inactivate, several repressors of DNA synthesis. The proportion of charged to uncharged tRNAs functions as a critical sensor of amino acid deficiency in both prokaryotes and eukaryotes (though through different mechanisms), mediating transcription and translation to improve the metabolic efficiency of amino acid synthesis. Also, uncharged tRNAs can act as ribozymes for the self-deletion of introns, again fostering increased metabolic efficiency under stress conditions. In addition, tRNA transcript fragments averaging about 30 to 35 nucleotides in length have been demonstrated to play diverse cellular roles, mainly in controlling transcription and translation in both prokaryotes and eukaryotes [
55]. These tRNA fragments are widespread and numerous in prokaryotic [
56] (and eukaryotic cells [
57] under stress (see also [
58,
59,
60,
61,
62]). tRNA fragments not only interact with other short RNA to regulate their activity, but also sequester RNA-binding proteins, buffering the translation system. Some tRNA-like repeats encode proteins that function specifically as Rho-dependent transcriptional regulators [
55]. Finally, eukaryotic tRNA genes sometimes function as “chromatin insulators”, separating active chromatin domains from silenced ones [
58]. Thus, tRNA and its fragments play important metabolic roles in replication, transcription and translation beyond simply recognizing specific codons on mRNAs and providing amino acids for ligation into proteins.
tRNA-derived small RNA (tsRNA) or tRNA fragments (tRF) have also been identified as novel regulatory RNAs controlling a wide range of physiological and pathological processes in organisms from Archaea through plants and animals [
63,
64,
65,
66]. tsRNA and tRF functions that have been identified include transcription regulation, cell proliferation, tumor genesis, stress response, intergenerational epigenetic inheritance, mRNA binding to ribosomes, amino acylation of tRNAs by amino acyl transferases, ribosomal protein function, and protection against retrotransposons such as SINEs [
67,
68,
69,
70].
Finally, tRNA participate in non-canonical forms of peptide synthesis independent of the ribosome, in conjunction with non-ribosomal peptide synthases. To quote Giessen and Marahiel, tRNA, “are also used as substrates and amino acid donors in a variety of other important cellular processes, ranging from bacterial cell wall biosynthesis, and lipid modification to protein turnover and secondary metabolite assembly” [
71]. For example, tRNA
Gly [
72] is involved in peptidoglycan synthesis, while tRNA
Lys participates in remodeling lipids [
73]. Ribosome-free production of cyclic dipeptides (diketopiperazines), for example, results in molecules that are essential for iron chelation or other types of siderophore activity; quorum sensing in bacteria; anti-viral, anti-bacterial, or anti-fungal activity, thereby protecting organisms from their pathogens [
74,
75].
Notably, tRNA may also have provided the material from which rRNA evolved. 5S rRNA has significant homologies with tRNA, and probably evolved directly from one or more tRNA precursors [
9,
10,
11,
76]. Like tRNAs, the 5S rRNA has also evolved to take on broader cellular functions. The 5S rRNA has not only undergone an alternative splicing event that has exonized it within the gene of its own transcription regulator, TFIIIA (transcription factor for polymerase III A) in all land plants [
77,
78], but has also mutated into an RNA mimic that regulates the alternative splicing of TFIIIA [
79]. The 5S rRNA can also exert extra-ribosomal function by binding to ribosomal proteins prior to their integration into functional ribosome complexes. One classic example is the control of the oncogene p53 by a 5S rRNA–RPL5–RPL11 complex that inhibits the Hdm2 protein that normally blocks p53 activity [
80,
81].
Like tRNA fragments, rRNA fragments (rRF) have also been found to exert a range of extra-ribosomal functions (
Table 2). For example, Arkov, et al. [
82] and Chernyaeva, et al. [
83], “identified a fragment of the
Escherichia coli 23S rRNA (nucleotides 74 to 136) whose expression caused readthrough of UGA nonsense mutations (but not UAA or UAG) in certain codon contexts in vivo. The antisense RNA fragment produced a similar effect…. Since termination at UGA in
E. coli specifically requires release factor 2 (RF2), our data suggest that the fragments interfere with RF2-dependent termination” [
83]. Similarly, Feagen, et al. [
84] reported high levels of rRNA fragments generated as functionally active transcripts from rDNA in
Plasmodium. Chen, et al., [
85] report that, “Two series of rRFs (rRF5 and rRF3) were precisely aligned to the 5′ and 3′ ends of the 5.8S and 28S rRNA genes. The rRF5 and rRF3 series were significantly more highly expressed than the rRFs located in the body of the rRNA genes. These series contained perfectly aligned reads, the lengths of which varied progressively, with 1-bp differences. The rRF5 and rRF3 series in the same expression pattern exist ubiquitously from ticks to human. The cellular experiments showed the RNAi knockdown of one 20-nt rRF3 induced the cell apoptosis and inhibited the cell proliferation.” Shi, et al. [
86] have developed a program called “SPORTS1.0”, for annotating and profiling additional non-coding RNAs optimized for rRNA- and tRNA-derived small RNAs.
rRNA fragments can also function as interference RNAs. Hadjiargyrou and Delihas [
87] have found that a large proportion of non-coding RNAs with regulatory functions derive from SINE elements, which are themselves often derived from tRNA-like sequences. Thus, tRNA and rRNA fragments appear to have fundamental regulatory roles in cellular metabolism that predate the origins of cellular life and have been incorporated as essential components of cellular life. Cerutti and Casas-Mollano [
88] suggest that interference functions for RNA were already in place prior to the origins of cellular life, making rRF and tRNA-derived small RNA fundamental aspects of ribosome function and evolution: “We have examined the taxonomic distribution and the phylogenetic relationship of key components of the RNA interference (RNAi) machinery in members of five eukaryotic supergroups. On the basis of the parsimony principle, our analyses suggest that a relatively complex RNAi machinery was already present in the last common ancestor of eukaryotes and consisted, at a minimum, of one Argonaute-like polypeptide, one Piwi-like protein, one Dicer, and one RNA-dependent RNA polymerase. As proposed before, the ancestral (but non-essential) role of these components may have been in defense responses against genomic parasites such as transposable elements and viruses. From a mechanistic perspective, the RNAi machinery in the eukaryotic ancestor may have been capable of both small-RNA-guided transcript degradation as well as transcriptional repression, most likely through histone modifications.”
2.5. Non-Canonical Functions of Aminoacyl tRNA Synthetases (aaRS)
As noted in the previous section, yet another class of ribosome-related proteins that have important additional cellular functions are the aminoacyl tRNA synthetases. The function usually ascribed to these proteins is to conjugate the appropriate amino acid to the correct tRNA. We have previously demonstrated that key functional elements of tRNA synthetases are encoded in all six reading frames of rRNAs, so that ribosomes may have had the ability to produce both tRNAs and the core enzyme structures that are required to load them with the correct amino acids [
6,
8]. Caprara, et al. [
113] and Mohr, et al. [
114] reported that the
Neurospora crassa mitochondrial (mt) tyrosyl-tRNA synthetase (CYT-18 protein) functions in splicing group I introns, in addition to aminoacylating tRNA(Tyr). Leucyl-tRNA synthetase has a similar function [
115,
116]. Lysyl-tRNA synthetase catalyzes the synthesis of dinucleoside polyphosphates in
E. coli [
117,
118] while the threonyl-tRNA synthetase from the same organism exerts autogenous control on its own mRNA, to self-regulate its translation [
119]. Other documented functions include the regulation of transcription, translation, splicing, inflammation (sometimes by mimicking cytokine activity) [
120,
121,
122], angiogenesis and apoptosis [
123], and cell signaling [
124,
125]. Some aaRSs interact specifically with other proteins [
126]. For example, many of these functions are mediated in higher eukaryotes by an aaRS complex involving eight different aaRS and three aminoacyl-tRNA synthetase-interacting multifunctional proteins (AIMS). The complex is formed of ArgRS, GlnRS, IleRS, LeuRS, LysRS, MetRS, AspRS, and Glu-ProRS around a core of AIMP1, AIMP2, and AIMP3, also known as p43, p38, and p18. The release of individual proteins from the complex triggers specific cell signaling activities (see TABLE) [
125,
127]. aaRS can also catalyze the synthesis of dinucleoside oligophosphates, and thus indirectly influence many other cell functions such as metal chelation and cell wall metabolism.
Evolutionary divergence from aa-synthetases has provided additional functions for “paralogs”, which resemble the aa-synthetases in sequence and structure, and sometimes also in function. These paralog functions have been reviewed by Pang, et al. [
127] and Schimmel, et al., [
128]. “For instance, the
E. coli YadB protein resembles GluRS and attaches glutamate to a queuosine base (position 34) at the first anticodon position of tRNA
Asp [
129]. This modification expands and alters the tRNA decoding of codons [
129,
130]. HisZ, a paralog of HisRS, is one component of the enzyme catalyzing the first step of histidine biosynthesis (Sissler, et al., 1999” [
131]. Like some of the tRNA synthetases, paralogs also carry out cyclodipeptide synthase activities [
132,
133].
Paralogs also play critical roles in translation by replacing missing amino acid tRNA synthetases. It is a common misconception is that all living organisms have a complete complement of 20 aaRS corresponding to each of the essential amino acids. In fact, 59% of organisms have less than the required 20, and 22% have more than the required 20, leaving only 19% with the exact complement that is often stated in textbooks. In organisms with both too few and too many aaRS, function is supplied by paralogs that are often able to modify one amino acid into another [
134]. Many of these paralogs have taken on additional, non-ribosomal functions that are summarized in
Table 7, which include amino acid synthesis, metabolic synthesis, polymerase activity, and stress and immunological functions [
128,
135,
136,
137,
138]. Notably, the human 50S ribosomal protein L2 and
Mycobacterium lysyl-tRNA synthetase are very similar [
139], suggesting that ribosomal proteins and tRNA synthetases may have common origins—another observation that is consistent with a ribosome-first origin of cellular function, but which could also emerge from a protein-first theory as well.
2.7. Theory Testing: How Well Do These Data Fit Other Possible Explanations of Genome Evolution?
Obviously, the data summarized above argues strongly for rRNA-containing genetic information that is expressed as translated proteins that are unrelated to ribosome function, and that rRNA-like and tRNA-like genetic sequences occur far more frequently than is either necessary or expected if rRNA and tRNA contain only structural, but no genetic, information. These finding raise the question of how much more frequent is such rRNA-like genetic information than one would expect. Here we attempt to address this question by comparing the frequency with which rRNA-like sequences appear in the E. coli K12 genome, as compared with the frequency that one would expect on the basis of a set of randomly generated rRNA-like sequences, and on the basis of a set of protein-encoding mRNAs of equivalent lengths.
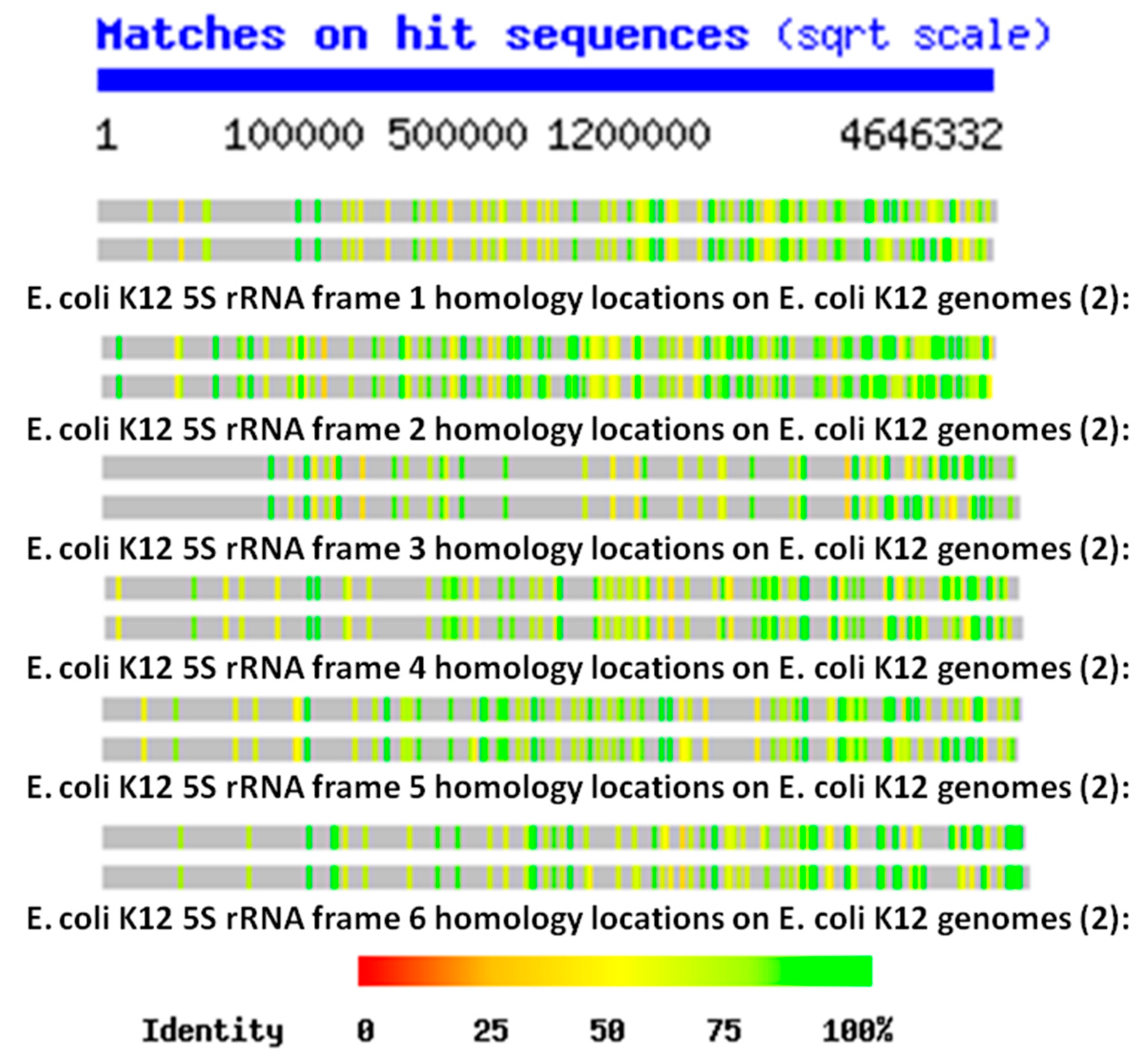
The key data for testing these three possibilities are
Figure 1 and
Figure 2, showing the distributions of rRNA-encoded proteins from all reading frames that are present in the
E. coli K12 genome. These figures show that, on average, the
E. coli K12 genome contains hundreds of copies of some version (translated in the six possible reading frames of the RNAs) of each of the 5S, 16S and 23S rRNAs. This observation is confirmed in the Tables below as well, and the data are consistent with the findings of Mauro and Edelman (1999) and many other studies of the ubiquity of rRNA-like sequences in the expressed proteins reviewed above. Four notable features of the locations of the rRNA homologies emerge from these plots. One is that the homology locations differ quite obviously between the 5S, 16S, and 23S homologies, indicating that they probably have independent origins (
Figure 1). Secondly, the locations also differ from one reading frame to another (
Figure 2), demonstrating that the homology search program is not merely picking up different versions of the same sequence. Third, the locations of the homologies differ even between closely related strains of
E. coli K12 (
Figure 1 and
Figure 2), indicating that they are subject to rearrangement processes. Finally, the homology locations, especially when averaged over all six possible reading frames, are more or less randomly distributed across the entire
E. coli genome so that any region of 10,000 or so base pairs is very likely to have several sequences homologous to at least one of the six reading frames of the 5S, 16S, or 23S rRNAs.
The data resulting from a comparison of the frequency and qualities of rRNA in the
E. coli genome confirm that ribosomal RNA-like sequences appear in the genome at far higher frequencies with much longer and more similar matches than do matches to randomized RNA sequences of equal length, or matches from
E. coli protein mRNA of equal length or randomized RNA sequences.
Table 8,
Table 9 and
Table 10 and
Figure 3 summarize the data comparing the frequency and quality of 5S rRNA homologies within the
E. coli K12 genome with data from a randomly selected set of protein mRNA of equivalent length and a set of randomly generated RNA sequences.
Table 11,
Table 12 and
Table 13 and
Figure 4 summarize the data comparing the frequency and quality of 16S rRNA homologies within the
E. coli K12 genome with data from a randomly selected set of protein mRNA of equivalent length, and a set of randomly generated RNA sequences.
Table 14,
Table 15 and
Table 16 and
Figure 5 summarize the data comparing the frequency and quality of 23S rRNA homologies within the
E. coli K12 genome with data from a randomly selected set of protein mRNA of equivalent length and a set of randomly generated RNA sequences.
In general (with the exception of the 23S protein matches) the number of rRNA homologies was significantly greater than the number of mRNA and random RNA matches. The number of matches differed by sample type (protein control, random control, or rRNA) for the 5S rRNA (ANOVA, F2,7 = 18.7,
p = 0.002), for the 16S (ANOVA, F
2,9 = 17.76,
p = 0.001), and for the 23S (ANOVA, F
2,6 = 187,
p = 2 × 10
−5). However, for the 23S samples, it was the protein control that had the greatest number of matches (see
Figure 5). The random RNA sequences had no identical matches with the
E. coli genome, where “identity” is defined as at least 90% identical amino acids over a sequence comprising at least 90% of the length of the RNA used as the search string. Thus, a 120 base pair (5S rRNA) would have to match a continuous sequence of 108 base pairs, with at least 90% identical amino acids in the
E. coli genome, to satisfy the “identity” classification. Each protein-derived mRNA sequence had one (and occasionally two) identities with the genome, as would be expected from the fact that these proteins are encoded in the genome. The rRNA sequences, in contrast, had dozens of identical matches scattered throughout the genome, as shown in
Figure 1 and
Figure 2. These data argue against a random or protein origin of the genome while supporting an rRNA origin.
Additional evidence for an rRNA origin was found in the quality measures derived from the homology searches. With the exception of the 5S rRNA matches, which were no longer on average than those derived from mRNAs or random RNA sequences, rRNA matches were significantly longer than the controls (
Figure 3,
Figure 4 and
Figure 5). Mean identity length did not differ across sample type (protein control, random control, or rRNA) for the 5S rRNA (ANOVA, F2,7 = 2.938,
p = 0.129). Mean identity length differed significantly by sample type for 16S rRNA (ANOVA, F
2,9 = 26.09,
p = 0.0003), and for 23S (ANOVA, F
2,6 = 97,253,
p = 3.35 × 10
−12).
Moreover, the number of identical amino acids found within these longer matches was also significantly greater, so that these longer matches were also better matches (
Figure 3,
Figure 4 and
Figure 5). The number of perfect identities differed by sample type (protein control, random control, or rRNA) for the 5S rRNA (ANOVA, F2,7 = 20,545,
p = 3.11 × 10
−12), 16S (ANOVA, F
2,9 = 3070,
p = 5.4 × 10
−11), and 23S (ANOVA, F
2,6 = 1.5 × 10
33,
p < 2 × 10
−16). The data derived by looking at amino acid similarities rather than identities was virtually identical from a statistical perspective; adding the similar amino acids within each sequence to the identities merely increased the quality of the matches without further differentiating the rRNAs from the controls, and so these data have not been displayed.
The average length of the rRNA matches is seven times that of the random RNA matches, and the number of identical amino acids within those matches is 14 times as many as in the random RNA matches. The mean length of identities in matches differed by sample type (protein control, random control, or rRNA) for the 5S rRNA (ANOVA, F2,7 = 23.54, p = 0.001), for 16S, (ANOVA, F2,9 = 1470, p = 5.43 × 10−11), and for 23S (ANOVA, F2,6 = 24,496, p = 1.05 × 10−10). Altogether, the rRNA matches account for 11 (16S rRNA) to 19 (23S rRNA) times the amount of the E. coli genome that is recognized as similar, as do the random RNA matches or the protein-derived mRNA matches. rRNA sequences are clearly highly over-represented compared with either control group.
In sum, rRNA sequences are significantly more likely to match the genome than are sets of mRNA derived from E. coli proteins of similar lengths. The average length of an rRNA match is six times that of an mRNA match (for 16S- and 23S-length sequences) and, as with the random sequences, the rRNA matches are significantly more likely than the mRNA matches to encode more amino acid identities so that overall the rRNAs encode four to six times as many identical amino acids within their matches (and this, in spite of the fact that BLAST yielded three times as many mRNA matches than 23S rRNA matches). Most significantly, while a typical mRNA sequence had 95% or more identity over its entire length (an “identity”) with only one genome sequence (as would be expected for matching its gene), the rRNA sequences yielded dozens of exact homologies. Thus, while protein sequences generally occur in only one copy in one reading frame, rRNA sequences occur in many copies in multiple reading frames. No random sequence in our study yielded any exact homologies. Protein mRNA matches are more similar to random sequence matches than to rRNA matches.
Overall, this statics-based similarity investigation into the frequency and qualities of rRNA-like, mRNA-like and randomized RNA-like sequences in the E. coli genome confirms that rRNA-like sequences occur very significantly more often than one would expect by chance, and far more often than mRNA duplications do. Indeed, it is possible to calculate the total number of base pairs involved in the similarities revealed in the Tables above, which yields the surprising result that, although the 5S, 16S, and 23S rRNAs, which total 4566 bp and account for only 0.098% of the 4,646,332 bp of the E. coli K12 genome, similar sequences (at E = 10 in the BLAST parameters) are present in sufficient copies (in all six reading frames) to account for 1/20 or 5% of the genome (240,171 bp) with quite high fidelity. This compares with 1/250 or 0.4% of the genome matching randomized RNA sequences (18,702 bp) and about 1/70 or 1.3% matching random mRNA (protein-encoding) sequences (66,768 bp), but with far less fidelity than for the rRNA sequences in terms of percent identities within each match. Thus, our hypothesis that rRNA may have been the basis for the first cellular genomes is supported while alternative theories such as a protein-world origin for the genome, or genome-before-ribosome, are not.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}