Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy


Abstract
:1. Introduction
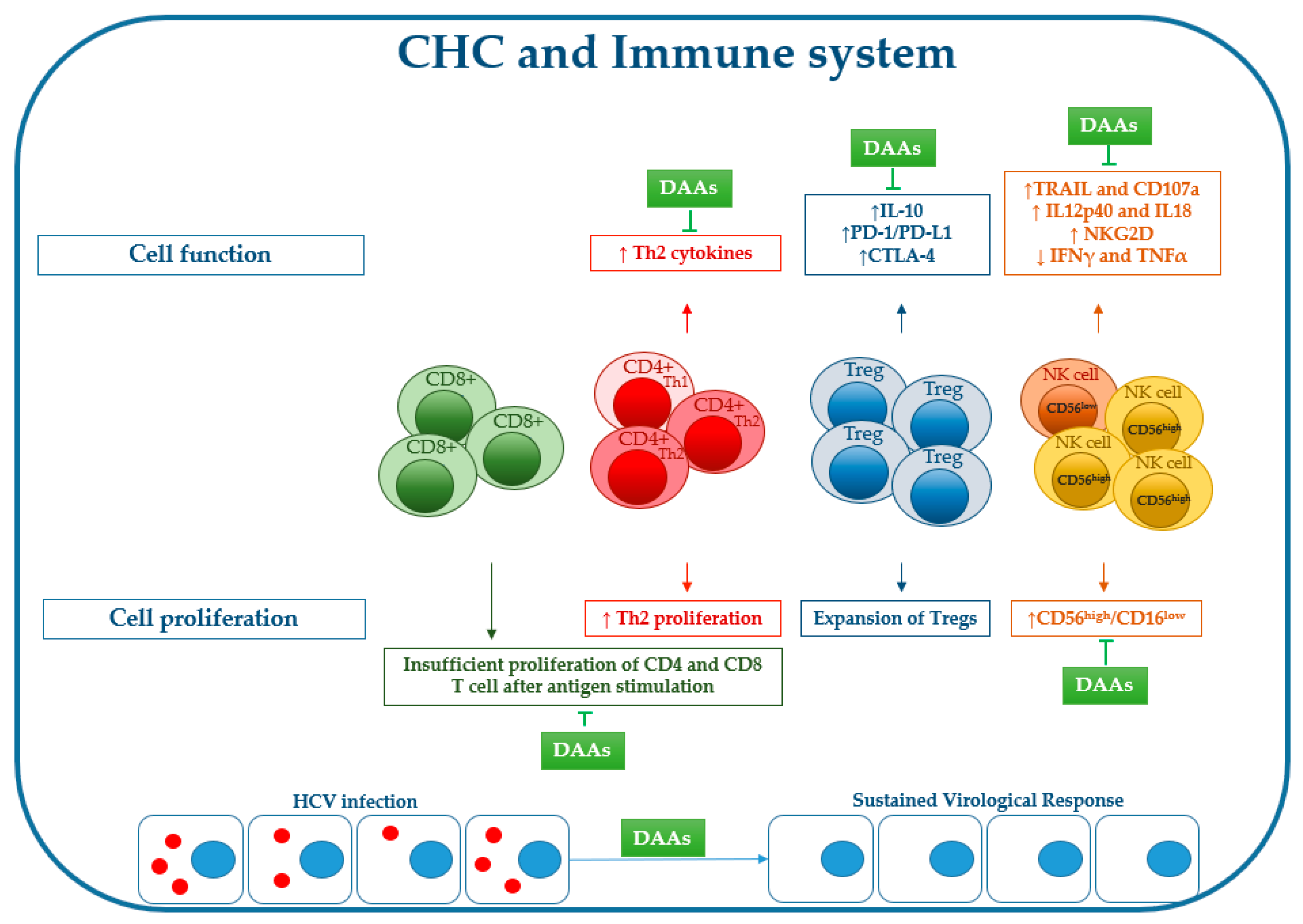
2. Immune Cell Dysfunction during Chronic HCV Infection
3. DAAs Are Able to Modulate Immune Cell Response
4. Cytokine Network Imbalance during Chronic C Hepatitis: Effect of DAAs
5. Potential Effect of DAAs in the Modulation of Angiogenesis Signaling
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| CHC | chronic hepatitis C |
| NS3 | nonstructural protein 3 |
| NS5A | nonstructural protein 5A |
| TNFα | tumor necrosis factor-alpha |
| NS5B | nonstructural protein 5B |
| RB1 | retinoblastoma-associated protein |
| MAVS | mitochondrial antiviral signaling protei |
| NK | natural killer |
| SVR | sustained virologic response |
| DAAs | direct-acting antivirals |
| IL12p40 | p40 subunit of IL12 |
| Treg | regulatory T cell |
| VEGF | vascular endothelial growth factor |
| IFN | interferon |
| TLR | toll-like receptor |
| IFNα | interferon alpha |
| TGFβ | transforming growth factor beta |
| PD-L1 | programmed death-ligand1 |
| CTLA-4 | cytotoxic Tl-lymphocyte associated protein 4 |
| IFNγ | interferon gamma |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| PD-1 | programmed death 1 |
| Ang-2 | angiopoietin |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| IL-2 | interlukin 2 |
| IL-4 | interlukin 4 |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IL-18 | interleukin 18 |
References
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 2001, 5, 87–107. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, I.I.; Tillmann, T.; Banerjee, A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Bialecki, E.S.; Di Bisceglie, A.M. Clinical presentation and natural course of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2005, 17, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Blum, H.E. Pathogenesis of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2005, 17, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S108–S111. [Google Scholar] [CrossRef] [PubMed]
- Farinati, F.; Cardin, R.; Bortolami, M.; Burra, P.; Russo, F.P.; Rugge, M.; Guido, M.; Sergio, A.; Naccarato, R. Hepatitis C virus: From oxygen free radicals to hepatocellular carcinoma. J. Viral Hepat. 2007, 14, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Fuchs, B.C.; Bardeesy, N.; Baumert, T.F.; Chung, R.T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 2014, 61, S79–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemel, R.; Gerechet, S.; Greif, H.; Bachmatove, L.; Birk, Y.; Golan-Goldhirsh, A.; Kunin, M.; Berdichevsky, Y.; Benhar, I.; Tur-Kaspa, R. Cell transformation induced by hepatitis C virus NS3 serine protease. J. Viral Hepat. 2001, 8, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Arima, N.; Kao, C.Y.; Licht, T.; Padmanabhan, R.; Sasaguri, Y.; Padmanabhan, R. Modulation of cell growth by the hepatitis C virus nonstructural protein NS5A. J. Biol. Chem. 2001, 276, 12675–12684. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Zhou, Y.; Kawai, S.; Eguchi, H.; Wands, J.R.; Li, J. Hepatitis C virus core protein stimulates hepatocyte growth: Correlation with upregulation of wnt-1 expression. Hepatology 2005, 41, 1096–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisi, A.; Giambartolomei, S.; Cupelli, F.; Merlo, P.; Fontemaggi, G.; Spaziani, A.; Balsano, C. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene 2003, 22, 2573–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Majumder, M.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein protects against TNF-α mediated apoptotic cell death. Virus Res. 2000, 67, 173–178. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.T.; Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Park, S.H.; Jang, K.L. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.L.; Baack, B.; Smith, B.D.; Yartel, A.; Pitasi, M.; Falck-Ytter, Y. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: A meta-analysis of observational studies. Ann. Intern. Med. 2013, 158, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bourliere, M.; Bronowicki, J.P.; de Ledinghen, V.; Hezode, C.; Zoulim, F.; Mathurin, P.; Tran, A.; Larrey, D.G.; Ratziu, V.; Alric, L.; et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: A randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect. Dis. 2015, 15, 397–404. [Google Scholar] [CrossRef]
- Leroy, V.; Angus, P.; Bronowicki, J.P.; Dore, G.J.; Hezode, C.; Pianko, S.; Pol, S.; Stuart, K.; Tse, E.; McPhee, F.; et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: A randomized phase III study (ALLY-3+). Hepatology 2016, 63, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Poordad, F.; Hezode, C.; Trinh, R.; Kowdley, K.V.; Zeuzem, S.; Agarwal, K.; Shiffman, M.L.; Wedemeyer, H.; Berg, T.; Yoshida, E.M.; et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N. Engl. J. Med. 2014, 370, 1973–1982. [Google Scholar] [CrossRef]
- Belli, L.S.; Berenguer, M.; Cortesi, P.A.; Strazzabosco, M.; Rockenschaub, S.R.; Martini, S.; Morelli, C.; Donato, F.; Volpes, R.; Pageaux, G.P.; et al. Delisting of liver transplant candidates with chronic hepatitis C after viral eradication: A European study. J. Hepatol. 2016, 65, 524–531. [Google Scholar] [CrossRef]
- Sievert, W.; Razavi, H.; Estes, C.; Thompson, A.J.; Zekry, A.; Roberts, S.K.; Dore, G.J. Enhanced antiviral treatment efficacy and uptake in preventing the rising burden of hepatitis C-related liver disease and costs in Australia. J. Gastroenterol. Hepatol. 2014, 29 (Suppl. S1), 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, K.; Moser, S.; Schwarzer, R.; Laferl, H.; Al-Zoairy, R.; Stauber, R.; Stattermayer, A.F.; Beinhardt, S.; Graziadei, I.; Freissmuth, C.; et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J. Hepatol. 2016, 65, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.; Vale, A.M.; Rodrigues, S.; Goncalves, R.; Albuquerque, A.; Pereira, P.; Lopes, S.; Silva, M.; Andrade, P.; Morais, R.; et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. J. Hepatol. 2016, 65, 1070–1071. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.M.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Aqel, B.A.; Pungpapong, S.; Gores, G.J.; Roberts, L.R.; Leise, M.D. Direct acting antiviral therapy and tumor recurrence after liver transplantation for hepatitis C-associated hepatocellular carcinoma. J. Hepatol. 2016, 65, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Zavaglia, C.; Okolicsanyi, S.; Cesarini, L.; Mazzarelli, C.; Pontecorvi, V.; Ciaccio, A.; Strazzabosco, M.; Belli, L.S. Is the risk of neoplastic recurrence increased after prescribing direct-acting antivirals for HCV patients whose HCC was previously cured? J. Hepatol. 2017, 66, 236–237. [Google Scholar] [CrossRef]
- Minami, T.; Tateishi, R.; Nakagomi, R.; Fujiwara, N.; Sato, M.; Enooku, K.; Nakagawa, H.; Asaoka, Y.; Kondo, Y.; Shiina, S.; et al. The impact of direct-acting antivirals on early tumor recurrence after radiofrequency ablation in hepatitis C-related hepatocellular carcinoma. J. Hepatol. 2016, 65, 1272–1273. [Google Scholar] [CrossRef]
- Nault, J.C.; Colombo, M. Hepatocellular carcinoma and direct acting antiviral treatments: Controversy after the revolution. J. Hepatol. 2016, 65, 663–665. [Google Scholar] [CrossRef]
- Meissner, E.G.; Wu, D.; Osinusi, A.; Bon, D.; Virtaneva, K.; Sturdevant, D.; Porcella, S.; Wang, H.; Herrmann, E.; McHutchison, J.; et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J. Clin. Investig. 2014, 124, 3352–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchill, M.A.; Golden-Mason, L.; Wind-Rotolo, M.; Rosen, H.R. Memory re-differentiation and reduced lymphocyte activation in chronic HCV-infected patients receiving direct-acting antivirals. J. Viral Hepat. 2015, 22, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Spaan, M.; van Oord, G.; Kreefft, K.; Hou, J.; Hansen, B.E.; Janssen, H.L.; de Knegt, R.J.; Boonstra, A. Immunological Analysis During Interferon-Free Therapy for Chronic Hepatitis C Virus Infection Reveals Modulation of the Natural Killer Cell Compartment. J. Infect. Dis. 2016, 213, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Casadei Gardini, A.; Conti, F.; Foschi, F.G.; Brillanti, S.; Andreone, P.; Mazzella, G.; Ravaioli, F. Imbalance of Neutrophils and Lymphocyte Counts Can Be Predictive of Hepatocellular Carcinoma Occurrence in Hepatitis C-related Cirrhosis Treated With Direct-acting Antivirals. Gastroenterology 2018, 154, 2281–2282. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.S.; Nakamoto, N.; Taniki, N.; Ojiro, K.; Amiya, T.; Makita, Y.; Murata, H.; Yamaguchi, A.; Shiba, S.; Miyake, R.; et al. On-treatment decrease of NKG2D correlates to early emergence of clinically evident hepatocellular carcinoma after interferon-free therapy for chronic hepatitis C. PLoS ONE 2017, 12, e0179096. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Li, Y.T.; Chen, Y.M.; Zhang, Y.; Zeng, Y.F.; Lin, C.S. Dynamic Changes of the Frequency of Classic and Inflammatory Monocytes Subsets and Natural Killer Cells in Chronic Hepatitis C Patients Treated by Direct-Acting Antiviral Agents. Can. J. Gastroenterol. Hepatol. 2017, 2017, 3612403. [Google Scholar] [CrossRef]
- Meissner, E.G.; Kohli, A.; Higgins, J.; Lee, Y.J.; Prokunina, O.; Wu, D.; Orr, C.; Masur, H.; Kottilil, S. Rapid changes in peripheral lymphocyte concentrations during interferon-free treatment of chronic hepatitis C virus infection. Hepatol. Commun. 2017, 1, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Langhans, B.; Nischalke, H.D.; Kramer, B.; Hausen, A.; Dold, L.; van Heteren, P.; Huneburg, R.; Nattermann, J.; Strassburg, C.P.; Spengler, U. Increased peripheral CD4(+) regulatory T cells persist after successful direct-acting antiviral treatment of chronic hepatitis C. J. Hepatol. 2017, 66, 888–896. [Google Scholar] [CrossRef]
- Villani, R.; Facciorusso, A.; Bellanti, F.; Tamborra, R.; Piscazzi, A.; Landriscina, M.; Vendemiale, G.; Serviddio, G. DAAs Rapidly Reduce Inflammation but Increase Serum VEGF Level: A Rationale for Tumor Risk during Anti-HCV Treatment. PLoS ONE 2016, 11, e0167934. [Google Scholar] [CrossRef]
- Debes, J.D.; van Tilborg, M.; Groothuismink, Z.M.A.; Hansen, B.E.; Schulze Zur Wiesch, J.; von Felden, J.; de Knegt, R.J.; Boonstra, A. Levels of Cytokines in Serum Associate With Development of Hepatocellular Carcinoma in Patients With HCV Infection Treated With Direct-Acting Antivirals. Gastroenterology 2018, 154, 515–517. [Google Scholar] [CrossRef]
- Carlin, A.F.; Aristizabal, P.; Song, Q.; Wang, H.; Paulson, M.S.; Stamm, L.M.; Schooley, R.T.; Wyles, D.L. Temporal dynamics of inflammatory cytokines/chemokines during sofosbuvir and ribavirin therapy for genotype 2 and 3 hepatitis C infection. Hepatology 2015, 62, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Faillaci, F.; Marzi, L.; Critelli, R.; Milosa, F.; Schepis, F.; Turola, E.; Andreani, S.; Vandelli, G.; Bernabucci, V.; Lei, B.; et al. Liver Angiopoietin-2 is a key predictor of de novo or recurrent hepatocellular cancer after HCV direct-acting antivirals. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Koziel, M.J. Cellular immune responses against hepatitis C virus. Clin. Infect. Dis. 2005, 41 (Suppl. S1), S25–S31. [Google Scholar] [CrossRef] [PubMed]
- Terilli, R.R.; Cox, A.L. Immunity and hepatitis C: A review. Curr. HIV/AIDS Rep. 2013, 10, 51–58. [Google Scholar] [CrossRef]
- Rigopoulou, E.I.; Zachou, K.; Gatselis, N.; Koukoulis, G.K.; Dalekos, G.N. Autoimmune hepatitis in patients with chronic HBV and HCV infections: Patterns of clinical characteristics, disease progression and outcome. Ann. Hepatol. 2013, 13, 127–135. [Google Scholar] [PubMed]
- Strassburg, C.P.; Vogel, A.; Manns, M.P. Autoimmunity and hepatitis C. Autoimmun. Rev. 2003, 2, 322–331. [Google Scholar] [CrossRef]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15661–15668. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Binder, M.; Bartenschlager, R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 2012, 36, 663–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.C.; Park, S.H.; Demino, M.; Nascimbeni, M.; Mihalik, K.; Major, M.; Veerapu, N.S.; Heller, T.; Feinstone, S.M.; Rice, C.M.; et al. Delayed induction, not impaired recruitment, of specific CD8(+) T cells causes the late onset of acute hepatitis C. Gastroenterology 2011, 141, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Thimme, R. T cell responses in hepatitis C: The good, the bad and the unconventional. Gut 2012, 61, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. USA 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Erickson, A.L.; Adams, E.J.; Kansopon, J.; Weiner, A.J.; Chien, D.Y.; Houghton, M.; Parham, P.; Walker, C.M. Analysis of a successful immune response against hepatitis C virus. Immunity 1999, 10, 439–449. [Google Scholar] [CrossRef]
- Takaki, A.; Wiese, M.; Maertens, G.; Depla, E.; Seifert, U.; Liebetrau, A.; Miller, J.L.; Manns, M.P.; Rehermann, B. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 2000, 6, 578–582. [Google Scholar] [CrossRef]
- Diepolder, H.M.; Gerlach, J.T.; Zachoval, R.; Hoffmann, R.M.; Jung, M.C.; Wierenga, E.A.; Scholz, S.; Santantonio, T.; Houghton, M.; Southwood, S.; et al. Immunodominant CD4+ T-cell epitope within nonstructural protein 3 in acute hepatitis C virus infection. J. Virol. 1997, 71, 6011–6019. [Google Scholar]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef]
- Missale, G.; Bertoni, R.; Lamonaca, V.; Valli, A.; Massari, M.; Mori, C.; Rumi, M.G.; Houghton, M.; Fiaccadori, F.; Ferrari, C. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Investig. 1996, 98, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Avia, M.; Martin, V.; Sevilla, N. IL-10: A Multifunctional Cytokine in Viral Infections. J. Immunol. Res. 2017, 2017, 6104054. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Teyton, L.; Oldstone, M.B.; McGavern, D.B. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 2005, 79, 10514–10527. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Tu, Z.; Soldevila-Pico, C.; Abdelmalek, M.; Zhu, H.; Xu, Y.L.; Cabrera, R.; Liu, C.; Davis, G.L. Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatology 2003, 38, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.L.; Liaw, Y.F.; Chen, M.H.; Huang, C.Y.; Kuo, G.C. Detection of type 2-like T-helper cells in hepatitis C virus infection: Implications for hepatitis C virus chronicity. Hepatology 1997, 25, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Tuma, R.A.; Pamer, E.G. Homeostasis of naive, effector and memory CD8 T cells. Curr. Opin. Immunol. 2002, 14, 348–353. [Google Scholar] [CrossRef]
- Piazzolla, G.; Tortorella, C.; Schiraldi, O.; Antonaci, S. Relationship between interferon-gamma, interleukin-10, and interleukin-12 production in chronic hepatitis C and in vitro effects of interferon-alpha. J. Clin. Immunol. 2000, 20, 54–61. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Mediating compromises between host and parasite. Nat. Immunol. 2003, 4, 10–11. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef]
- Piccirillo, C.A.; Shevach, E.M. Cutting edge: Control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J. Immunol. 2001, 167, 1137–1140. [Google Scholar] [CrossRef]
- Billerbeck, E.; Bottler, T.; Thimme, R. Regulatory T cells in viral hepatitis. World J. Gastroenterol. 2007, 13, 4858–4864. [Google Scholar] [CrossRef]
- Barjon, C.; Dahlqvist, G.; Calmus, Y.; Conti, F. Role of regulatory T-cells during hepatitis C infection: From the acute phase to post-transplantation recurrence. Dig. Liver Dis. 2015, 47, 913–917. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wan, L.; Zhang, C.; Zheng, X.; Li, J.; Chen, Z.K. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology 2009, 214, 342–349. [Google Scholar] [CrossRef]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clemenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS ONE 2013, 8, e83139. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Chiu, J.; Ernst, D.M.; Keating, A. Acquired Natural Killer Cell Dysfunction in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Front. Immunol. 2018, 9, 267. [Google Scholar] [CrossRef]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef]
- Serti, E.; Chepa-Lotrea, X.; Kim, Y.J.; Keane, M.; Fryzek, N.; Liang, T.J.; Ghany, M.; Rehermann, B. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology 2015, 149, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009, 137, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, G.; Titerence, R.H.; Koh, C.; Edlich, B.; Feld, J.J.; Rotman, Y.; Ghany, M.G.; Hoofnagle, J.H.; Liang, T.J.; Heller, T.; et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 2010, 138, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, M.U. Direct-Acting Antivirals Cure Innate Immunity in Chronic Hepatitis C. Gastroenterology 2015, 149, 25–28. [Google Scholar] [CrossRef]
- Missale, G.; Pilli, M.; Zerbini, A.; Penna, A.; Ravanetti, L.; Barili, V.; Orlandini, A.; Molinari, A.; Fasano, M.; Santantonio, T.; et al. Lack of full CD8 functional restoration after antiviral treatment for acute and chronic hepatitis C virus infection. Gut 2012, 61, 1076–1084. [Google Scholar] [CrossRef]
- Martin, B.; Hennecke, N.; Lohmann, V.; Kayser, A.; Neumann-Haefelin, C.; Kukolj, G.; Bocher, W.O.; Thimme, R. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J. Hepatol. 2014, 61, 538–543. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Bedard, N.; Badr, G.; Ostrowski, M.; Sekaly, R.P.; Bruneau, J.; Willems, B.; Heathcote, E.J.; Shoukry, N.H. Comparison of immune restoration in early versus late alpha interferon therapy against hepatitis C virus. J. Virol. 2010, 84, 10429–10435. [Google Scholar] [CrossRef]
- Seigel, B.; Bengsch, B.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Thimme, R. Factors that determine the antiviral efficacy of HCV-specific CD8(+) T cells ex vivo. Gastroenterology 2013, 144, 426–436. [Google Scholar] [CrossRef]
- Ahlenstiel, G. The natural killer cell response to HCV infection. Immune Netw. 2013, 13, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Novick, D.; Rubinstein, M.; Siegmund, B.; Enrich, B.; Koch, R.O.; Vogel, W.; Kim, S.H.; Dinarello, C.A.; Tilg, H. Interferon-alpha induces interleukin-18 binding protein in chronic hepatitis C patients. Clin. Exp. Immunol. 2002, 129, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; O’Garra, A.; de Waal Malefyt, R.; Vieira, P.; Mosmann, T.R. Interleukin-10. Annu. Rev. Immunol. 1993, 11, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef] [Green Version]
- Snell, L.M.; Osokine, I.; Yamada, D.H.; De la Fuente, J.R.; Elsaesser, H.J.; Brooks, D.G. Overcoming CD4 Th1 Cell Fate Restrictions to Sustain Antiviral CD8 T Cells and Control Persistent Virus Infection. Cell Rep. 2016, 16, 3286–3296. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Flynn, J.K.; Dore, G.J.; Hellard, M.; Yeung, B.; Rawlinson, W.D.; White, P.A.; Kaldor, J.M.; Lloyd, A.R.; Ffrench, R.A.; Group, A.S. Early IL-10 predominant responses are associated with progression to chronic hepatitis C virus infection in injecting drug users. J. Viral Hepat. 2011, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- De Maria, A.; Fogli, M.; Mazza, S.; Basso, M.; Picciotto, A.; Costa, P.; Congia, S.; Mingari, M.C.; Moretta, L. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur. J. Immunol. 2007, 37, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockman, M.A.; Kwon, D.S.; Tighe, D.P.; Pavlik, D.F.; Rosato, P.C.; Sela, J.; Porichis, F.; Le Gall, S.; Waring, M.T.; Moss, K.; et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 2009, 114, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Parish, I.A.; Marshall, H.D.; Staron, M.M.; Lang, P.A.; Brustle, A.; Chen, J.H.; Cui, W.; Tsui, Y.C.; Perry, C.; Laidlaw, B.J.; et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J. Clin. Investig. 2014, 124, 3455–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. The origin of FOXP3-expressing CD4+ regulatory T cells: Thymus or periphery. J. Clin. Investig. 2003, 112, 1310–1312. [Google Scholar] [CrossRef]
- Ulsenheimer, A.; Gerlach, J.T.; Gruener, N.H.; Jung, M.C.; Schirren, C.A.; Schraut, W.; Zachoval, R.; Pape, G.R.; Diepolder, H.M. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 2003, 37, 1189–1198. [Google Scholar] [CrossRef]
- MacDonald, A.J.; Duffy, M.; Brady, M.T.; McKiernan, S.; Hall, W.; Hegarty, J.; Curry, M.; Mills, K.H. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J. Infect. Dis. 2002, 185, 720–727. [Google Scholar] [CrossRef]
- Graham, C.S.; Wells, A.; Liu, T.; Sherman, K.E.; Peters, M.; Chung, R.T.; Bhan, A.K.; Andersen, J.; Koziel, M.J.; Team, A.S. Antigen-specific immune responses and liver histology in HIV and hepatitis C coinfection. AIDS 2005, 19, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.E.; Ikeda, F.; Li, Y.; Nakamoto, N.; Ganesan, S.; Valiga, M.E.; Nunes, F.A.; Rajender Reddy, K.; Chang, K.M. Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J. Hepatol. 2008, 48, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, M.; Sene, D.; Pol, S.; Bourliere, M.; Poynard, T.; Charlotte, F.; Cacoub, P.; Caillat-Zucman, S. Intrahepatic virus-specific IL-10-producing CD8 T cells prevent liver damage during chronic hepatitis C virus infection. Hepatology 2006, 44, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accapezzato, D.; Francavilla, V.; Paroli, M.; Casciaro, M.; Chircu, L.V.; Cividini, A.; Abrignani, S.; Mondelli, M.U.; Barnaba, V. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J. Clin. Investig. 2004, 113, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Oh, B.S.; Kwon, J.H.; You, C.R.; Chung, K.W.; Kay, C.S.; Jung, H.S. Serum interleukin-6 and C-reactive protein as a prognostic indicator in hepatocellular carcinoma. Cytokine 2012, 60, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Hu, X.; Liu, S.; He, B. Prognostic and Therapeutic Values of Tumor Necrosis Factor-Alpha in Hepatocellular Carcinoma. Med. Sci. Monit. 2016, 22, 3694–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrajoli, A.; Keating, M.J.; Manshouri, T.; Giles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002, 100, 1215–1219. [Google Scholar]
- Ahmed, M.I.; Salahy, E.E.; Fayed, S.T.; El-Hefnawy, N.G.; Khalifa, A. Human papillomavirus infection among Egyptian females with cervical carcinoma: Relationship to spontaneous apoptosis and TNF-alpha. Clin. Biochem. 2001, 34, 491–498. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Grimshaw, M.J.; Kulbe, H.; Wilson, J.L.; Wilbanks, G.D.; Burke, F.; Balkwill, F.R. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol. Cancer Ther. 2006, 5, 382–390. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Kono, H.; Amemiya, H.; Matsuda, M.; Suzuki, T.; Maki, A.; Fujii, H. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int. J. Cancer 2006, 118, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Stark, G.R. NFkappaB-dependent signaling pathways. Exp. Hematol. 2002, 30, 285–296. [Google Scholar] [CrossRef]
- Hassan, M.; Selimovic, D.; El-Khattouti, A.; Soell, M.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Megahed, M. Hepatitis C virus-mediated angiogenesis: Molecular mechanisms and therapeutic strategies. World J. Gastroenterol. 2014, 20, 15467–15475. [Google Scholar] [CrossRef] [PubMed]
- Tandle, A.; Blazer, D.G., III; Libutti, S.K. Antiangiogenic gene therapy of cancer: Recent developments. J. Transl. Med. 2004, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, X.; Medina, J.; Sanz-Cameno, P.; Garcia-Buey, L.; Martin-Vilchez, S.; Borque, M.J.; Lopez-Cabrera, M.; Moreno-Otero, R. The potential of angiogenesis soluble markers in chronic hepatitis C. Hepatology 2005, 42, 696–701. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Elpek, G.O. Angiogenesis and liver fibrosis. World J. Hepatol. 2015, 7, 377–391. [Google Scholar] [CrossRef]
- Duffy, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Vascular Endothelial Growth Factor (VEGF) and Its Role in Non-Endothelial Cells: Autocrine Signalling by VEGF. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Mise, M.; Arii, S.; Higashituji, H.; Furutani, M.; Niwano, M.; Harada, T.; Ishigami, S.; Toda, Y.; Nakayama, H.; Fukumoto, M.; et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996, 23, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Horbach, A.; Kubitz, R.; Frilling, A.; Haussinger, D. Disruption of hepatocellular tight junctions by vascular endothelial growth factor (VEGF): A novel mechanism for tumor invasion. J. Hepatol. 2004, 41, 274–283. [Google Scholar] [CrossRef]
- Park, Y.N.; Kim, Y.B.; Yang, K.M.; Park, C. Increased expression of vascular endothelial growth factor and angiogenesis in the early stage of multistep hepatocarcinogenesis. Arch. Pathol. Lab. Med. 2000, 124, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Bupathi, M.; Kaseb, A.; Janku, F. Angiopoietin 2 as a therapeutic target in hepatocellular carcinoma treatment: Current perspectives. OncoTargets Ther. 2014, 7, 1927–1932. [Google Scholar] [CrossRef]
- Torimura, T.; Ueno, T.; Kin, M.; Harada, R.; Taniguchi, E.; Nakamura, T.; Sakata, R.; Hashimoto, O.; Sakamoto, M.; Kumashiro, R.; et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 2004, 40, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Ker, C.G.; Chen, H.Y.; Juan, C.C.; Lo, H.W.; Shen, Y.Y.; Chen, J.S.; Lee, K.T.; Sheen, P.C. Role of angiogenesis in hepatitis and hepatocellular carcinoma. Hepatogastroenterology 1999, 46, 646–650. [Google Scholar] [PubMed]
- Garcia-Monzon, C.; Sanchez-Madrid, F.; Garcia-Buey, L.; Garcia-Arroyo, A.; Garcia-Sanchez, A.; Moreno-Otero, R. Vascular adhesion molecule expression in viral chronic hepatitis: Evidence of neoangiogenesis in portal tracts. Gastroenterology 1995, 108, 231–241. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Waris, G.; Mikolon, D.; Stupack, D.G.; Siddiqui, A. Hepatitis C virus stabilizes hypoxia-inducible factor 1alpha and stimulates the synthesis of vascular endothelial growth factor. J. Virol. 2007, 81, 10249–10257. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.K.; Brimacombe, C.L.; Rowe, I.A.; Reynolds, G.M.; Fletcher, N.F.; Stamataki, Z.; Bhogal, R.H.; Simoes, M.L.; Ashcroft, M.; Afford, S.C.; et al. A dual role for hypoxia inducible factor-1alpha in the hepatitis C virus lifecycle and hepatoma migration. J. Hepatol. 2012, 56, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.; Caveda, L.; Sanz-Cameno, P.; Arroyo, A.G.; Martin-Vilchez, S.; Majano, P.L.; Garcia-Buey, L.; Sanchez-Madrid, F.; Moreno-Otero, R. Hepatocyte growth factor activates endothelial proangiogenic mechanisms relevant in chronic hepatitis C-associated neoangiogenesis. J. Hepatol. 2003, 38, 660–667. [Google Scholar] [CrossRef]
- Mazzanti, R.; Messerini, L.; Monsacchi, L.; Buzzelli, G.; Zignego, A.L.; Foschi, M.; Monti, M.; Laffi, G.; Morbidelli, L.; Fantappie, O.; et al. Chronic viral hepatitis induced by hepatitis C but not hepatitis B virus infection correlates with increased liver angiogenesis. Hepatology 1997, 25, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.C.; Fisher, R.A. Angiogenesis soluble factors as hepatocellular carcinoma noninvasive markers for monitoring hepatitis C virus cirrhotic patients awaiting liver transplantation. Transplantation 2007, 84, 1262–1271. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
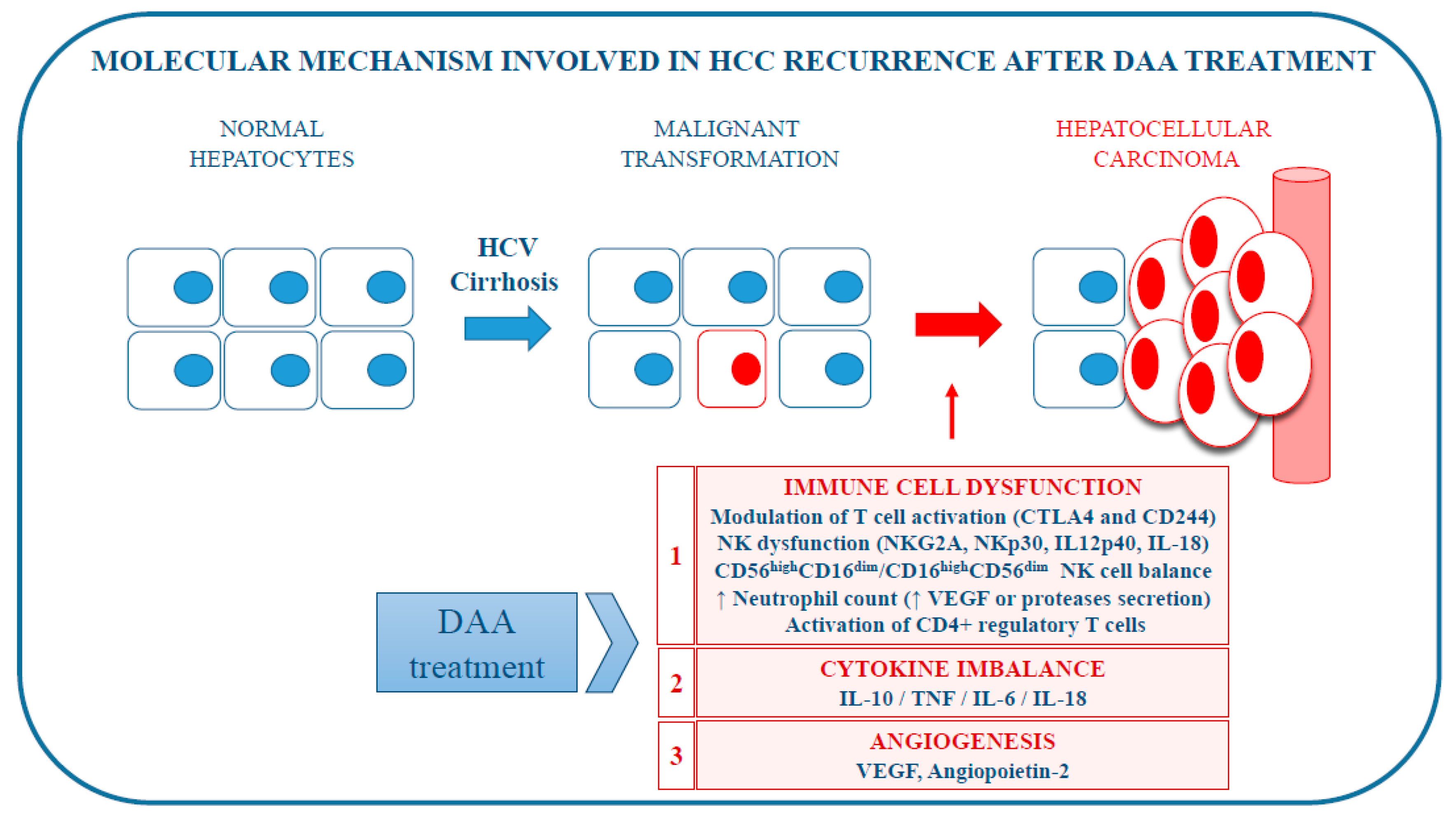
| Hypothesis | Molecular Mechanism | Author | Ref. |
|---|---|---|---|
| Immune cell dysfunction | Modulation of T cell activation | Meissner et al., 2014 | [41] |
| Reduced NK cell activation | Burchill et al., 2015 | [42] | |
| Downregulation of NKG2A receptor | Spaan et al., 2016 | [43] | |
| Declined IL12p40, IL-18 serum level | Spaan et al., 2016 | [43] | |
| Modulation of differential white blood cell count | Casadei Gardini et al., 2018 | [44] | |
| Imbalance in NK cell subgroups | Chu et al., 2017 | [45] | |
| Decreased NKG2D | Chu et al., 2017 | [45] | |
| Decreased frequencies of NK cells | Ning et al., 2017; | [46] | |
| Meissner et al., 2017 | [47] | ||
| Immunosuppressive Tregs function | Langhans et al., 2017 | [48] | |
| Change in immune cytokine network | Rapid reduction of IL-10 serum level | Villani et al., 2016 | [49] |
| Increased TNFα secretion | Debes et al., 2018 | [50] | |
| Change in IL-6 serum level | Debes et al., 2018 | [50] | |
| Change in IL-18 serum level | Spaan et al., 2016; | [43] | |
| Ning et al., 2017; | [46] | ||
| Carlin et al., 2015 | [51] | ||
| Activation of angiogenesis | Increase of VEGF serum level | Villani et al., 2016; | [49] |
| Faillaci et al., 2018 | [52] | ||
| Increase of angiopoietin-2 serum level | Faillaci et al., 2018 | [52] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villani, R.; Vendemiale, G.; Serviddio, G. Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. Int. J. Mol. Sci. 2019, 20, 49. https://doi.org/10.3390/ijms20010049
Villani R, Vendemiale G, Serviddio G. Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. International Journal of Molecular Sciences. 2019; 20(1):49. https://doi.org/10.3390/ijms20010049
Chicago/Turabian StyleVillani, Rosanna, Gianluigi Vendemiale, and Gaetano Serviddio. 2019. "Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy" International Journal of Molecular Sciences 20, no. 1: 49. https://doi.org/10.3390/ijms20010049
APA StyleVillani, R., Vendemiale, G., & Serviddio, G. (2019). Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. International Journal of Molecular Sciences, 20(1), 49. https://doi.org/10.3390/ijms20010049