MCEE Mutations in an Adult Patient with Parkinson’s Disease, Dementia, Stroke and Elevated Levels of Methylmalonic Acid

 ,
,
Abstract
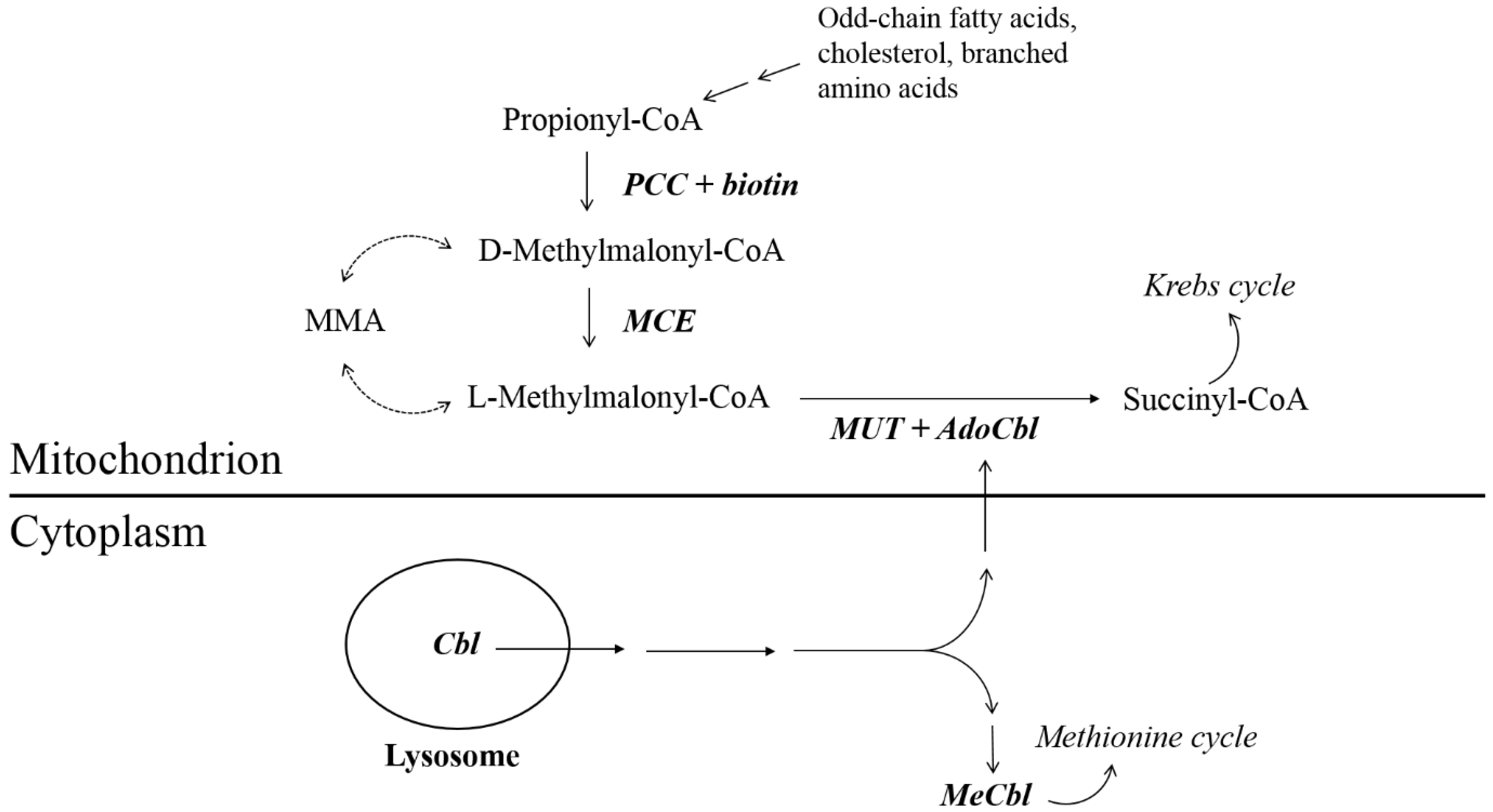
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hannibal, L.; Lysne, V.; Bjorke-Monsen, A.L.; Behringer, S.; Grunert, S.C.; Spiekerkoetter, U.; Jacobsen, D.W.; Blom, H.J. Biomarkers and algorithms for the diagnosis of vitamin b12 deficiency. Front. Mol. Biosci. 2016, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.; Rosenblatt, D.S. Inborn errors of cobalamin absorption and metabolism. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157C, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ktena, Y.P.; Paul, S.M.; Hauser, N.S.; Sloan, J.L.; Gropman, A.; Manoli, I.; Venditti, C.P. Delineating the spectrum of impairments, disabilities, and rehabilitation needs in methylmalonic acidemia (mma). Am. J. Med. Genet. A 2015, 167A, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Gradinger, A.; Longo, N.; Wu, X.; Leclerc, D.; Lerner-Ellis, J.; Lemieux, M.; Belair, C.; Watkins, D.; Rosenblatt, D.S.; et al. Homozygous nonsense mutation in the mcee gene and sirna suppression of methylmalonyl-coa epimerase expression: A novel cause of mild methylmalonic aciduria. Mol. Genet. Metab. 2006, 88, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.A.; Mamer, O.A.; Scriver, C.R. Metabolism of methylmalonic acid in rats. Is methylmalonyl-coenzyme a racemase deficiency symptomatic in man? J. Clin. Invest. 1983, 72, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Bikker, H.; Bakker, H.D.; Abeling, N.G.; Poll-The, B.T.; Kleijer, W.J.; Rosenblatt, D.S.; Waterham, H.R.; Wanders, R.J.; Duran, M. A homozygous nonsense mutation in the methylmalonyl-coa epimerase gene (mcee) results in mild methylmalonic aciduria. Hum. Mutat. 2006, 27, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Gradinger, A.B.; Belair, C.; Worgan, L.C.; Li, C.D.; Lavallee, J.; Roquis, D.; Watkins, D.; Rosenblatt, D.S. Atypical methylmalonic aciduria: Frequency of mutations in the methylmalonyl coa epimerase gene (mcee). Hum. Mutat. 2007, 28, 1045. [Google Scholar] [CrossRef] [PubMed]
- Waters, P.J.; Thuriot, F.; Clarke, J.T.; Gravel, S.; Watkins, D.; Rosenblatt, D.S.; Levesque, S. Methylmalonyl-coa epimerase deficiency: A new case, with an acute metabolic presentation and an intronic splicing mutation in the mcee gene. Mol. Genet. Metab. Rep. 2016, 9, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Abily-Donval, L.; Torre, S.; Samson, A.; Sudrié-Arnaud, B.; Acquaviva, C.; Guerrot, A.M.; Benoist, J.F.; Marret, S.; Bekri, S.; Tebani, A. Methylmalonyl-coa epimerase deficiency mimicking propionic aciduria. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, K.; Bailey, H.J.; Burda, P.; Chaikuad, A.; Krysztofinska, E.; Suormala, T.; Burer, C.; Lutz, S.; Fowler, B.; Froese, D.S.; et al. Genetic, structural, and functional analysis of pathogenic variations causing methylmalonyl-coa epimerase deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.K.; Guo, T.; Soukhova, N.; Soldin, S.J. Rapid measurement of plasma acylcarnitines by liquid chromatography-tandem mass spectrometry without derivatization. Clin. Chim. Acta 2005, 358, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Alm, J.; Hagenfeldt, L.; Larsson, A. Concentrations of organic acids in the urine of healthy newborn children. Ann. Clin. Biochem. 1978, 15, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Engvall, M.; Naess, K.; Lesko, N.; Larsson, P.; Dahlberg, M.; Andeer, R.; Wredenberg, A.; Freyer, C.; Barbaro, M.; et al. Rapid pulsed whole genome sequencing for comprehensive acute diagnostics of inborn errors of metabolism. BMC Genomics 2014, 15, 1090. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; IJlst, L.; Duran, M.; Mienie, L.J.; van Cruchten, A.; van der Westhuizen, F.H.; Wanders, R.J. Inhibition of n-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme a esters and the production of alternative glutamate esters. Biochim. Biophys. Acta 2014, 1842, 2510–2516. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Biochemical Analyses | 2005 Apr, Dec | 2011 Mar | 2013 Mar, Oct | 2015 Oct, Dec | 2016 Jan, Jun |
|---|---|---|---|---|---|
| MMA (µmol/l) [<0,40] | 8.1 a, 11 a | 180 a | -, 35 a | 9.9 b, 16 b,e | -, 18 b |
| p-Hcy (µmol/l) [5,0-15]c, [5,0-20]d | 17 c, 18 c | 26 d, 24 d,e | -, 32 c | ||
| s-cobalamin (pmol/l) [150-650] | 510, 460 | 420 | -, 410 | ||
| s-folate (nmol/l) [7-40] | >40, - | >40 | |||
| p-propionyl-carnitine (µmol/l) [<1,3] | -, 13 | ||||
| u-methylmalonate (mmol/mol creatinine) [<10] | 9.0, 5.5 | 10, 60 | |||
| p-creatinine (µmol/l) [<100] | -, 124 | 117 | 119, 133 | -, 189 | 159, - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andréasson, M.; Zetterström, R.H.; von Döbeln, U.; Wedell, A.; Svenningsson, P. MCEE Mutations in an Adult Patient with Parkinson’s Disease, Dementia, Stroke and Elevated Levels of Methylmalonic Acid. Int. J. Mol. Sci. 2019, 20, 2631. https://doi.org/10.3390/ijms20112631
Andréasson M, Zetterström RH, von Döbeln U, Wedell A, Svenningsson P. MCEE Mutations in an Adult Patient with Parkinson’s Disease, Dementia, Stroke and Elevated Levels of Methylmalonic Acid. International Journal of Molecular Sciences. 2019; 20(11):2631. https://doi.org/10.3390/ijms20112631
Chicago/Turabian StyleAndréasson, Mattias, Rolf H. Zetterström, Ulrika von Döbeln, Anna Wedell, and Per Svenningsson. 2019. "MCEE Mutations in an Adult Patient with Parkinson’s Disease, Dementia, Stroke and Elevated Levels of Methylmalonic Acid" International Journal of Molecular Sciences 20, no. 11: 2631. https://doi.org/10.3390/ijms20112631