Perspectives for Applying G-Quadruplex Structures in Neurobiology and Neuropharmacology

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
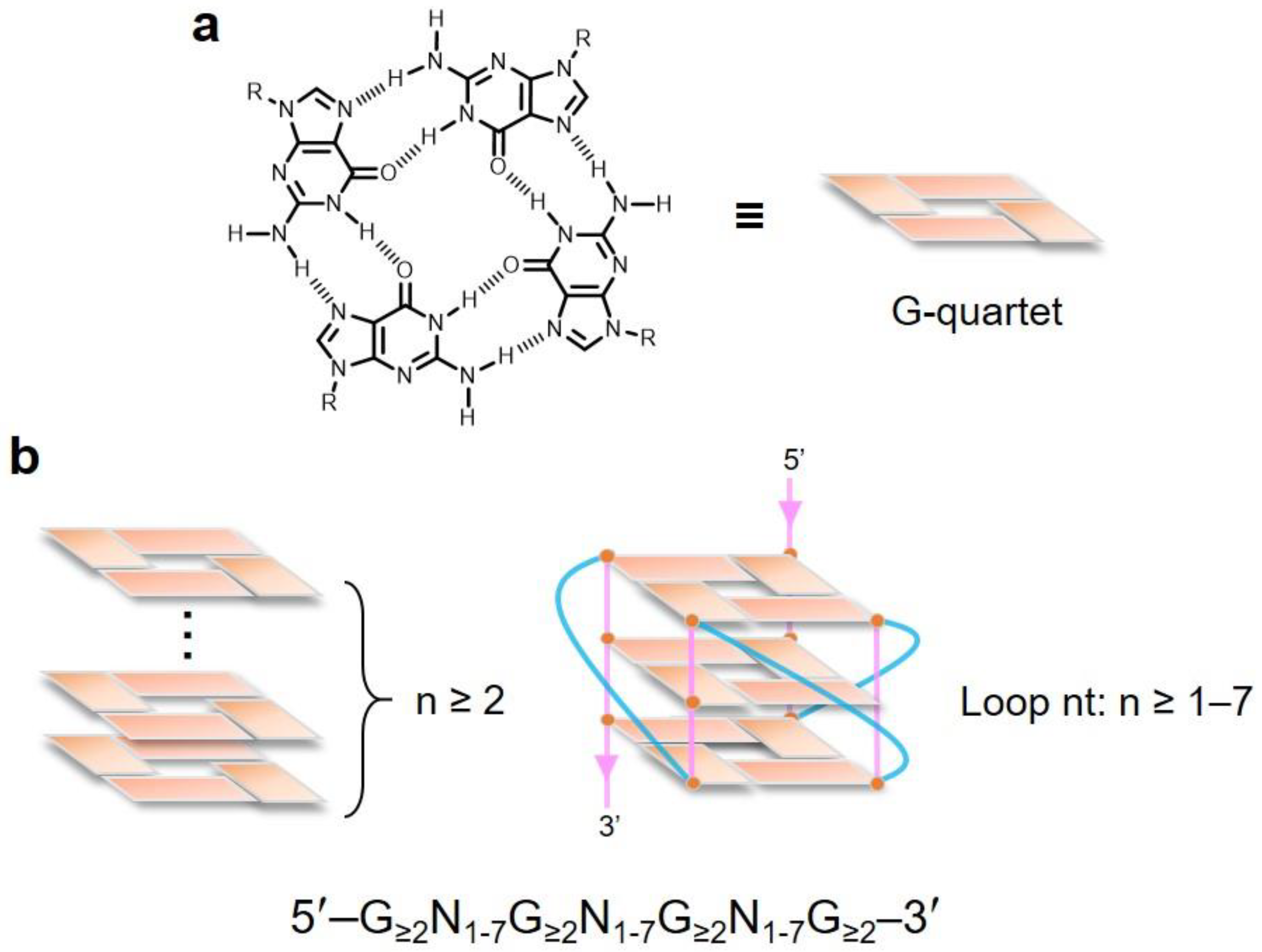
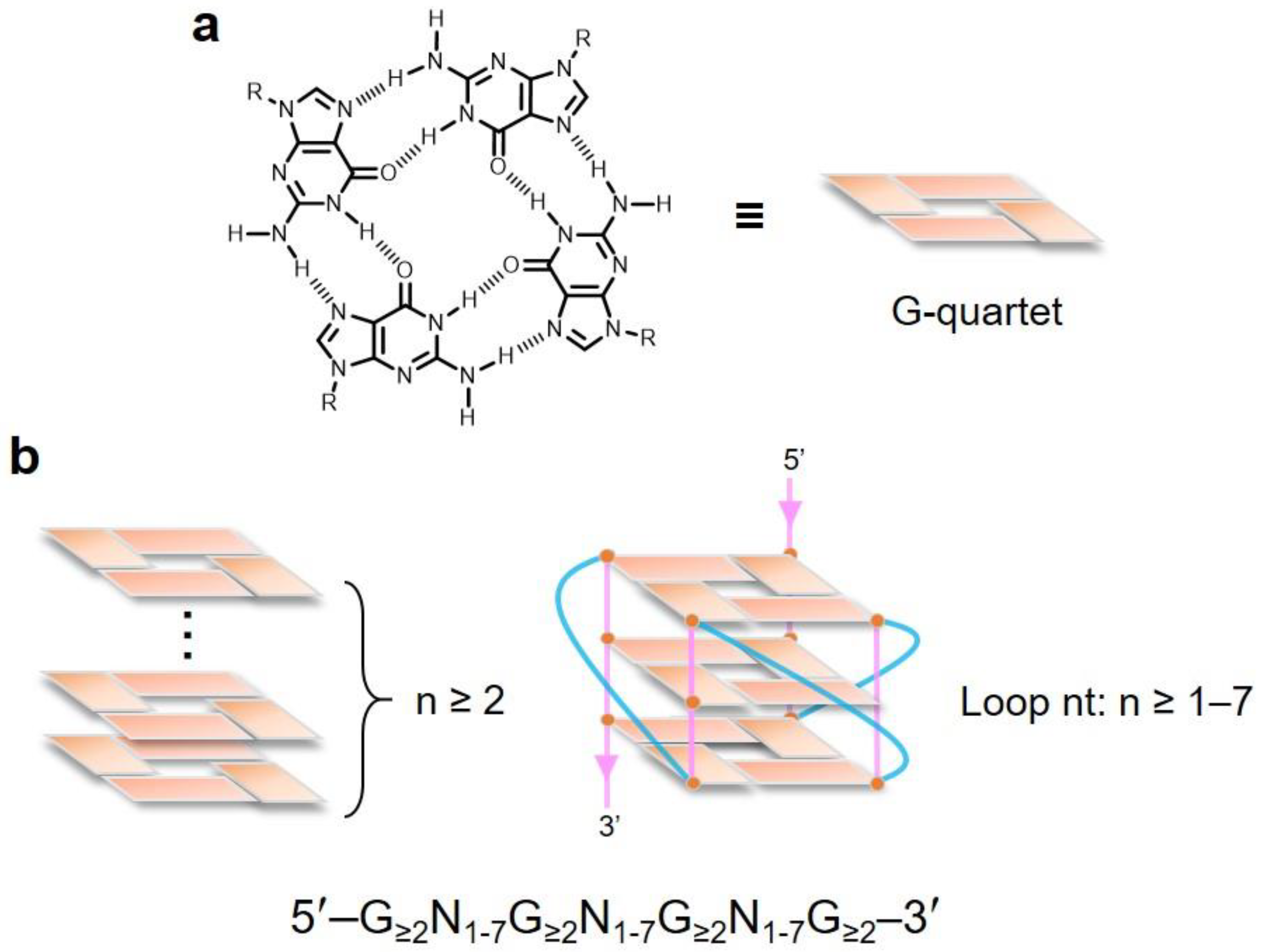
2. Topological Analysis of G-Quadruplex
3. Genome-Wide Analysis of G-Quadruplexes
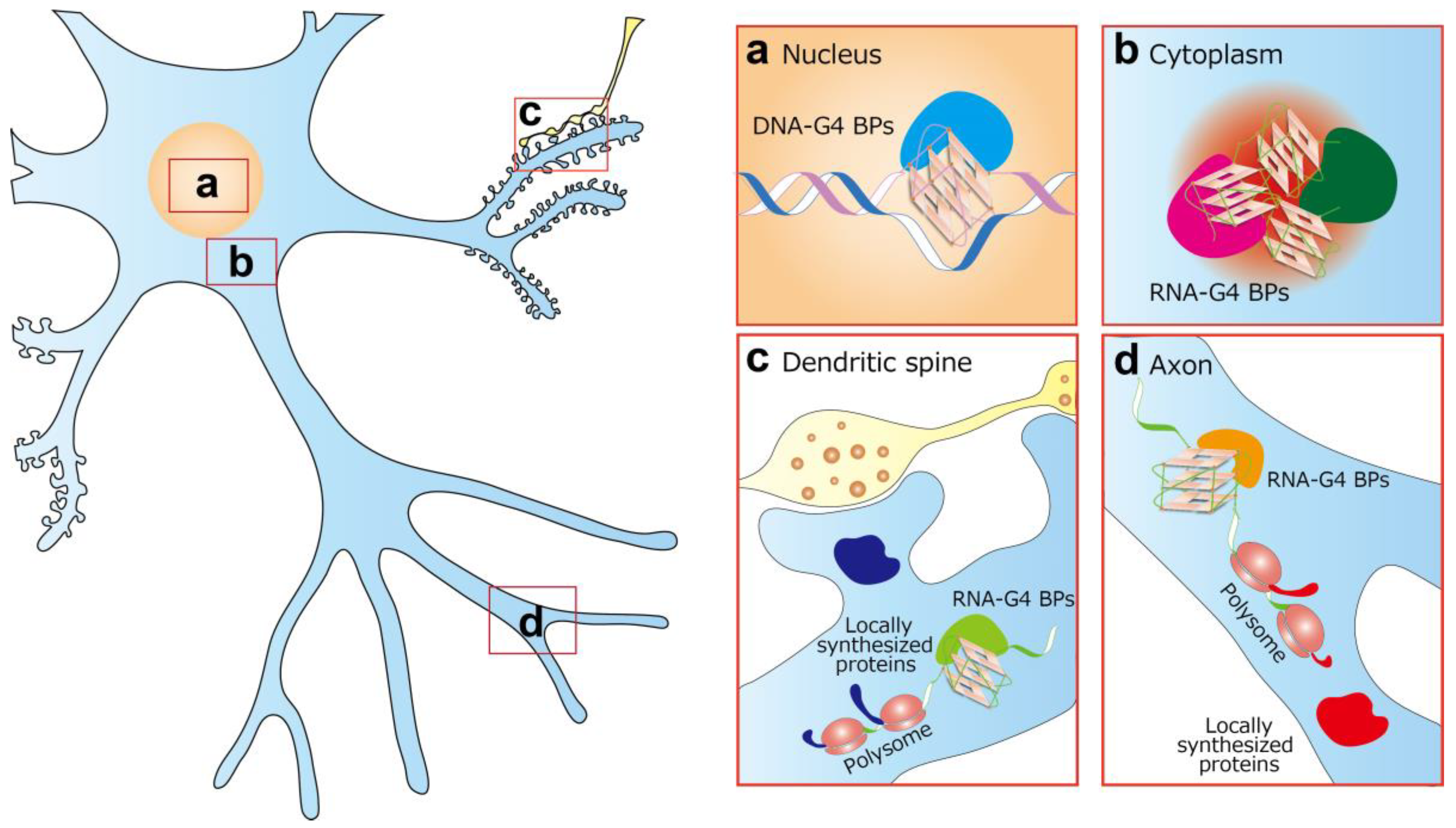
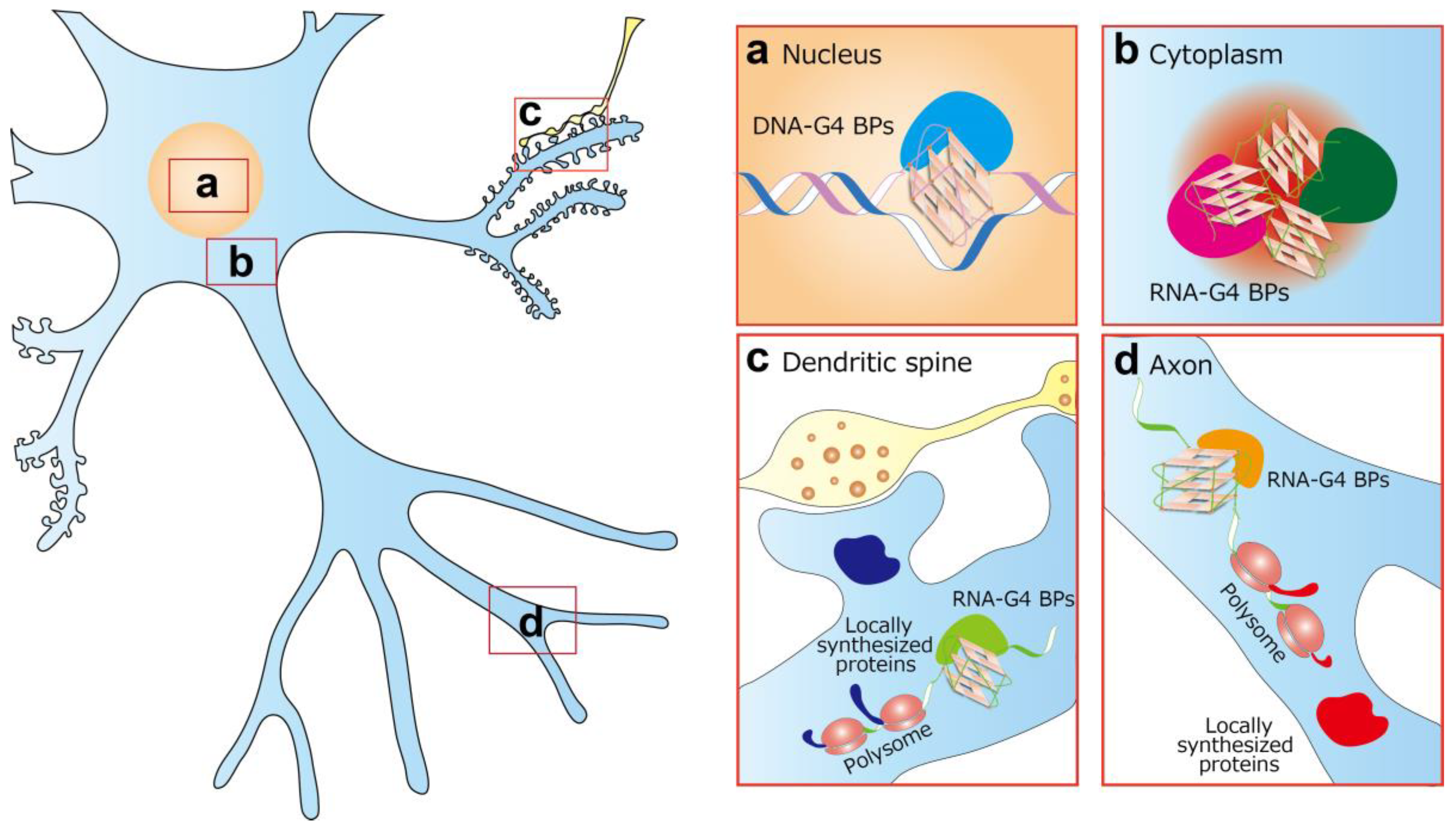
4. DNA-G4 and Neurological Diseases
5. RNA-G4 and Neurological Diseases
6. G-Quadruplex Is a Therapeutic Target for Neurological Diseases
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DNA-G4 | DNA G-quadruplex |
| RNA-G4 | RNA G-quadruplex |
| PQS | putative G-quadruplex sequences |
| UTR | untranslated region |
| ChIP-seq | chromatin immunoprecipitation-sequencing |
| RT | reverse transcriptase |
| HRE | hexanucleotide repeat expansion |
| RBP | RNA-binding protein |
| C9ALS/FTD | C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia |
| RAN | repeat associated non-AUG |
| ATR-X | X-linked alpha thalassemia intellectual disability |
| DNMT | DNA methyltransferase |
| CaMKIIα | calcium calmodulin-dependent protein kinase IIα |
| FXS | fragile X syndrome |
| 5-ALA | 5-aminolevulinic acid |
References
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Gehring, K.; Leroy, J.L.; Gueron, M. A tetrameric DNA-structure with protonated cytosine–cytosine base-pairs. Nature 1993, 363, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel 4-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Bang, I. Untersuchungen über die Guanylsäure. Biochem. Z. 1910, 26, 293–311. [Google Scholar]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef]
- Williamson, J.R.; Raghuraman, M.K.; Cech, T.R. Monovalent cation-induced structure of telomeric DNA: The G-Quartet model. Cell 1989, 59, 871–880. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Perreault, J.P.; Topisirovic, I.; Richard, S. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.M.; Lyons, S.M.; Ivanov, P. RNA G-quadruplexes in biology: Principles and molecular mechanisms. J. Mol. Biol. 2017, 429, 2127–2147. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Merrick, C.J. G-Quadruplexes: Prediction, characterization, and biological application. Trends Biotechnol. 2017, 35, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.K.; Sharma, S.; Chowdhury, S. Non-duplex G-quadruplex structures emerge as mediators of epigenetic modifications. Trends Genet. 2019, 35, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Braunlin, W.H. Duplex to quadruplex equilibrium of the self-complementary oligonucleotide d(GGGGCCCC). Biopolymers 1995, 35, 677–681. [Google Scholar] [CrossRef]
- Kumar, N.; Sahoo, B.; Varun, K.A.; Maiti, S.; Maiti, S. Effect of loop length variation on quadruplex-Watson Crick duplex competition. Nucleic Acids Res. 2008, 36, 4433–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, D.; Mirihana-Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front Chem. 2016, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiset Miserachs, H.; Donghi, D.; Börner, R.; Johannsen, S.; Sigel, R.K. Distinct differences in metal ion specificity of RNA and DNA G-quadruplexes. J. Biol. Inorg. Chem. 2016, 21, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.A. Quadruplex structures in nucleic acids. Biopolymers 2000, 56, 123–146. [Google Scholar] [CrossRef]
- Yaku, H.; Fujimoto, T.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Phthalocyanines: A new class of G-quadruplex-ligands with many potential applications. Chem. Commun. 2012, 48, 6203–6216. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Joachimi, A.; Benz, A.; Hartig, J.S. A comparison of DNA and RNA quadruplex structures and stabilities. Bioorg. Med. Chem. 2009, 17, 6811–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.Y.; Bugaut, A.; Balasubramanian, S. A sequence-independent analysis of the loop length dependence of intramolecular RNA G-quadruplex stability and topology. Biochemistry 2011, 50, 7251–7258. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- Eddy, J.; Maizels, N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.K.; Abraham, J.K.; Mani, P.; Kulshrestha, R.; Chowdhury, S. QuadBase: Genome-wide database of G4 DNA--occurrence and conservation in human, chimpanzee, mouse and rat promoters and 146 microbes. Nucleic Acids Res. 2008, 36, D381–D385. [Google Scholar] [CrossRef]
- Beaudoin, J.D.; Jodoin, R.; Perreault, J.P. New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res. 2014, 42, 1209–1223. [Google Scholar] [CrossRef]
- Bedrat, A.; Lacroix, L.; Mergny, J.L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.M.; Lemaitre, J.M. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bugaut, A.; Huppert, J.L.; Balasubramanian, S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007, 3, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, V.T.; Phan, A.T. Bulges in G-quadruplexes: Broadening the definition of G-quadruplex-forming sequences. J. Am. Chem. Soc. 2013, 135, 5017–5028. [Google Scholar] [CrossRef] [PubMed]
- Garant, J.M.; Perreault, J.P.; Scott, M.S. Motif independent identification of potential RNA G-quadruplexes by G4RNA screener. Bioinformatics 2017, 33, 3532–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garant, J.M.; Luce, M.J.; Scott, M.S.; Perreault, J.P. G4RNA: An RNA G-quadruplex database. Database 2015. [Google Scholar] [CrossRef] [PubMed]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 8, 877–881. [Google Scholar] [CrossRef]
- Yoshida, W.; Saikyo, H.; Nakabayashi, K.; Yoshioka, H.; Bay, D.H.; Iida, K.; Kawai, T.; Hata, K.; Ikebukuro, K.; Nagasawa, K.; et al. Identification of G-quadruplex clusters by high-throughput sequencing of whole-genome amplified products with a G-quadruplex ligand. Sci. Rep. 2018, 8, 3116. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 10, 1267–1272. [Google Scholar] [CrossRef]
- Kwok, C.K.; Marsico, G.; Sahakyan, A.B.; Chambers, V.S.; Balasubramanian, S. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 2016, 10, 841–844. [Google Scholar] [CrossRef]
- Guo, J.U.; Bartel, D.P. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, aaf5371. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Marsico, G.; Balasubramanian, S. Detecting RNA G-uadruplexes (rG4s) in the Transcriptome. Cold Spring Harb. Perspect. Biol. 2018, 10, a032284. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N. G4-associated human diseases. EMBO Rep. 2015, 16, 910–922. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., 3rd; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS 77. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Fratta, P.; Mizielinska, S.; Nicoll, A.J.; Zloh, M.; Fisher, E.M.C.; Parkinson, G.; Isaacs, A.M. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012, 2, 1016. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.M.; Anderson, P.J.; Ivanov, P. ALS/FTD-associated C9ORF72 repeat RNA promotes phase transitions in vitro and in cells. Cell Rep. 2017, 21, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Valton, A.L.; Prioleau, M.N. G-quadruplexes in DNA replication: A problem or a necessity? Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; Suthers, G.K.; Wilkie, A.O.; Buckle, V.J.; Higgs, D.R. X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: Localization to Xq12-q21.31 by X inactivation and linkage analysis. Am. J. Hum. Genet. 1992, 51, 1136–1149. [Google Scholar] [PubMed]
- Gibbons, R.J.; Picketts, D.J.; Villard, L.; Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Wada, T.; Fisher, C.A.; Malik, N.; Mitson, M.J.; Steensma, D.P.; Fryer, A.; Goudie, D.R.; Krantz, I.D.; Traeger-Synodinos, J. Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 2008, 29, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Argentaro, A.; Yang, J.C.; Chapman, L.; Kowalczyk, M.S.; Gibbons, R.J.; Higgs, D.R.; Neuhaus, D.; Rhodes, D. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. USA 2007, 104, 11939–11944. [Google Scholar] [CrossRef] [Green Version]
- Dhayalan, A.; Tamas, R.; Bock, I.; Tattermusch, A.; Dimitrova, E.; Kudithipudi, S.; Ragozin, S.; Jeltsch, A. The ATRX–ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 2011, 20, 2195–2203. [Google Scholar] [CrossRef]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Picketts, D.J.; Higgs, D.R.; Bachoo, S.; Blake, D.J.; Quarrell, O.W.; Gibbons, R.J. ATRX encodes a novel member of the SNF2 family of proteins: Mutations point a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 1996, 5, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Mitson, M.; Kelley, L.A.; Sternberg, M.J.; Higgs, D.R.; Gibbons, R.J. Functional significance of mutations in the Snf2 domain of ATRX. Hum. Mol. Genet. 2011, 20, 2603–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 2014, 159, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA antagonizes ATRX and protects telomeres. Cell 2017, 170, 86–101. [Google Scholar] [CrossRef]
- Kernohan, K.D.; Jiang, Y.; Tremblay, D.C.; Bonvissuto, A.C.; Eubanks, J.H.; Mann, M.R.; Bérubé, N.G. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 2010, 18, 191–202. [Google Scholar] [CrossRef]
- Kernohan, K.D.; Vernimmen, D.; Gloor, G.B.; Bérubé, N.G. Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 2014, 42, 8356–8368. [Google Scholar] [CrossRef]
- Shioda, N.; Beppu, H.; Fukuda, T.; Li, E.; Kitajima, I.; Fukunaga, K. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 2011, 31, 346–358. [Google Scholar] [CrossRef]
- Nogami, T.; Beppu, H.; Tokoro, T.; Moriguchi, S.; Shioda, N.; Fukunaga, K.; Ohtsuka, T.; Ishii, Y.; Sasahara, M.; Shimada, Y.; et al. Reduced expression of the ATRX gene, a chromatin-remodeling factor, causes hippocampal dysfunction in mice. Hippocampus 2011, 21, 678–687. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Yamaguchi, K.; Onozato, M.; Li, Y.; Kurosawa, K.; Tanabe, H.; Okamoto, N.; Era, T.; Sugiyama, H.; et al. Targeting G-quadruplex DNA as cognitive function therapy for ATR-X syndrome. Nat. Med. 2018, 24, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Higgs, D.R.; Gibbons, R.J. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, H. RNA reigns in neurons. Neurons 2005, 48, 1–6. [Google Scholar] [CrossRef]
- Sutton, M.A.; Schuman, E.M. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 2006, 127, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Wells, D.G. Dendritic mRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.C.; Ephrussi, A. mRNA localization: Gene expression in the spatial dimension. Cell 2009, 136, 719–730. [Google Scholar] [CrossRef]
- Subramanian, M.; Rage, F.; Tabet, R.; Flatter, E.; Mandel, J.L.; Moine, H. G–quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep. 2011, 12, 697–704. [Google Scholar] [CrossRef]
- Miller, S.; Yasuda, M.; Coats, J.K.; Jones, Y.; Martone, M.E.; Mayford, M. Disruption of dendritic translation of CaMKIIα impairs stabilization of synaptic plasticity and memory consolidation. Neuron 2002, 36, 507–519. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tawani, A.; Mishra, A.; Kumar, A. G4IPDB: A database for G-quadruplex structure forming nucleic acid interacting proteins. Sci. Rep. 2016, 6, 38144. [Google Scholar] [CrossRef] [Green Version]
- Ule, J. Ribonucleoprotein complexes in neurological diseases. Curr. Opin. Neurobiol. 2008, 18, 516–523. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Bassell, G.J. Fragile balance: RNA editing tunes the synapse. Nat. Neurosci. 2011, 14, 1492–1494. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.T.; Wilkinson, K.D.; Reines, D.; Warren, S.T. FMR1 protein: Conserved RNP family domains and selective RNA binding. Science 1993, 262, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Schaeffer, C.; Bardoni, B.; Mandel, J.L.; Ehresmann, B.; Ehresmann, C.; Moine, H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001, 20, 4803–4813. [Google Scholar] [CrossRef]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 1993, 74, 291–298. [Google Scholar] [CrossRef]
- Siomi, M.C.; Zhang, Y.; Siomi, H.; Dreyfuss, G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol. Cell. Biol. 1996, 16, 3825–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrick, L.K.; Hashimoto, H.; Cheng, X.; Warren, S.T. Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum. Mol. Genet. 2015, 24, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Darnell, J.C.; Serganov, A.; Majumdar, A.; Ilin, S.; Raslin, T.; Polonskaia, A.; Chen, C.; Clain, D.; et al. Structure-function studies of FMRP RGG peptide recognition of an RNA duplex-quadruplex junction. Nat. Struct. Mol. Biol. 2011, 18, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, N.; Polonskaia, A.; Darnell, J.C.; Darnell, R.B.; Patel, D.J.; Serganov, A. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. USA 2015, 112, E5391–E5400. [Google Scholar] [CrossRef] [PubMed]
- Laggerbauer, B.; Ostareck, D.; Keidel, E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wang, H.; Liang, Z.; Ku, L.; O’Donnell, W.T.; Li, W.; Warren, S.T.; Feng, Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. USA 2004, 101, 15201–15206. [Google Scholar] [CrossRef] [Green Version]
- Menon, L.; Mader, S.A.; Mihailescu, M.R. Fragile X mental retardation protein interactions with the microtubule associated protein 1B RNA. RNA 2008, 14, 1644–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westmark, C.J.; Malter, J.S. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007, 5, e52. [Google Scholar] [CrossRef]
- Castets, M.; Schaeffer, C.; Bechara, E.; Schenck, A.; Khandjian, E.W.; Luche, S.; Moine, H.; Rabilloud, T.; Mandel, J.L.; Bardoni, B. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum. Mol. Genet. 2005, 14, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, Y.; Stackpole, E.E.; Novak, A.; Gao, Y.; Zhao, Y.; Zhao, X.; Richter, J.D. Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, E11397–E11405. [Google Scholar] [CrossRef] [PubMed]
- Wieland, M.; Hartig, J.S. RNA quadruplex-based modulation of gene expression. Chem. Biol. 2007, 14, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Halder, K.; Wieland, M.; Hartig, J.S. Predictable suppression of gene expression by 5′-UTR-based RNA quadruplexes. Nucleic Acids Res. 2009, 37, 6811–6817. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Marsico, G.; Herdy, B.; Ghanbarian, A.T.; Portella, G.; Balasubramanian, S. RNA G-quadruplexes at upstream open reading frames cause DHX36- and DHX9-dependent translation of human mRNAs. Genome Biol. 2018, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Menon, L.; Mihailescu, M.R. Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res. 2007, 35, 5379–5392. [Google Scholar] [CrossRef] [Green Version]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent progress of targeted G-quadruplex-preferred ligands toward cancer therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Bando, T.; Sugiyama, H. Ligand design to acquire specificity to intended G-quadruplex structures. Chemistry 2019, 25, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Roberts, D.W.; Valdés, P.A.; Harris, B.T.; Hartov, A.; Fan, X.; Ji, S.; Leblond, F.; Tosteson, T.D.; Wilson, B.C.; Paulsen, K.D. Glioblastoma multiforme treatment with clinical trials for surgical resection (aminolevulinic acid). Neurosurg. Clin. N. Am. 2012, 23, 371–377. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014, 83, 1043–1050. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly (GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asamitsu, S.; Takeuchi, M.; Ikenoshita, S.; Imai, Y.; Kashiwagi, H.; Shioda, N. Perspectives for Applying G-Quadruplex Structures in Neurobiology and Neuropharmacology. Int. J. Mol. Sci. 2019, 20, 2884. https://doi.org/10.3390/ijms20122884
Asamitsu S, Takeuchi M, Ikenoshita S, Imai Y, Kashiwagi H, Shioda N. Perspectives for Applying G-Quadruplex Structures in Neurobiology and Neuropharmacology. International Journal of Molecular Sciences. 2019; 20(12):2884. https://doi.org/10.3390/ijms20122884
Chicago/Turabian StyleAsamitsu, Sefan, Masayuki Takeuchi, Susumu Ikenoshita, Yoshiki Imai, Hirohito Kashiwagi, and Norifumi Shioda. 2019. "Perspectives for Applying G-Quadruplex Structures in Neurobiology and Neuropharmacology" International Journal of Molecular Sciences 20, no. 12: 2884. https://doi.org/10.3390/ijms20122884