The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer


Abstract
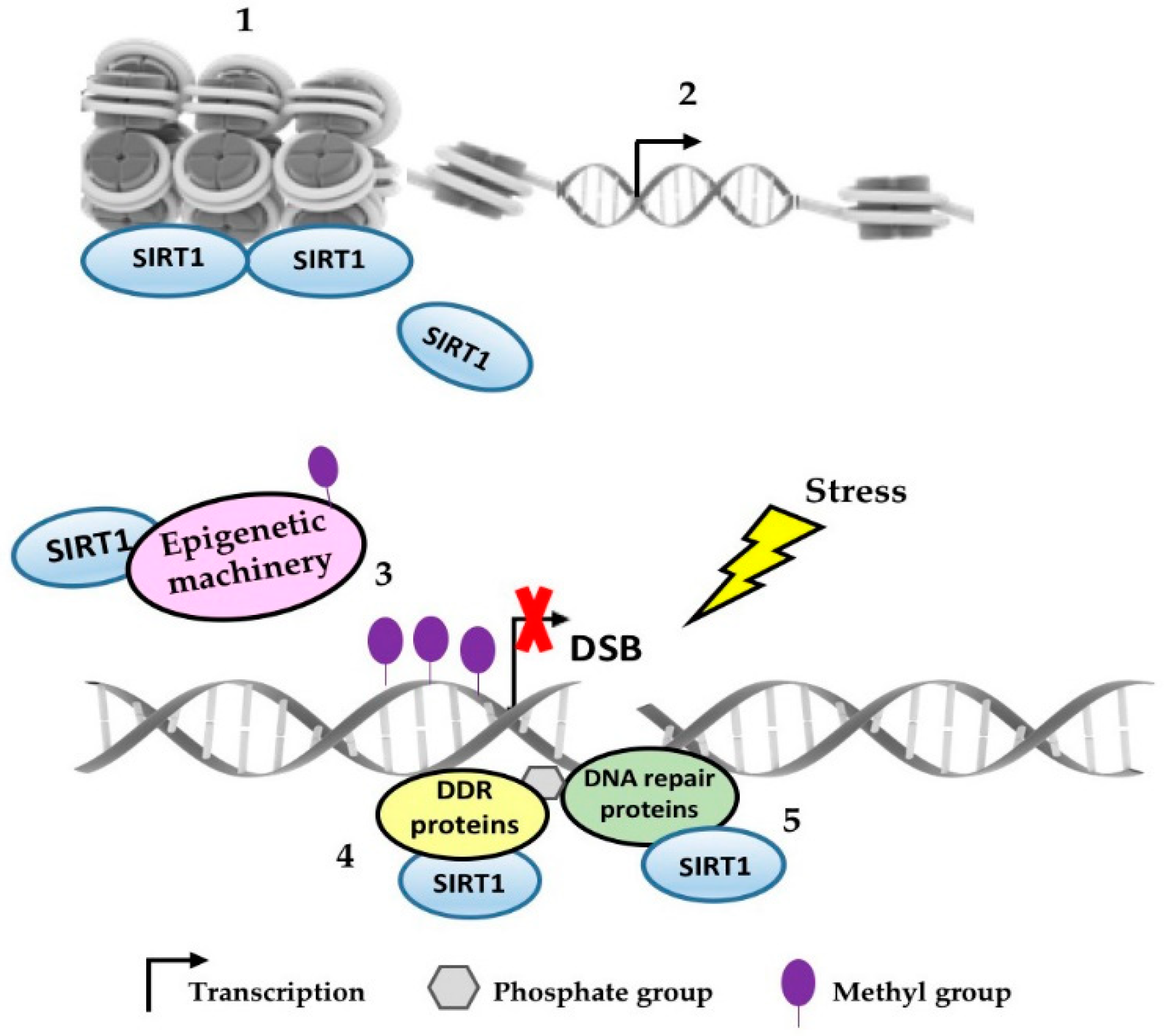
:1. Introduction
1.1. DNA Damage Signaling Response
1.2. Epigenetic Effects of DNA Damage
2. SIRT1 as an Epigenetic Regulator in Response to DNA Damage
3. Other Forms by which SIRT1 Modulates the DNA Repair Response
4. Concluding Remarks
Funding
Conflicts of Interest
References
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Thachootam, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug. Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Houtgraaf, J.H.; Versmissen, J.; Giessen, J.V. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med. 2006, 7, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Hoejimakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Huh, Y.J.; Cho, H.; Lee, B.; Park, J.; Hwang, D.-Y.; Kim, D.-W. SIRT1 enhances the survival of human embryonic stem cells by promoting DNA Repair. Stem. Cell Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christimann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lobrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Barnard, S.; Moquet, J. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef]
- Filippo, J.S.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Weterings, E.; Chen, D.J. The endless tale of non-homologous end-joining. Cell Res. 2008, 18, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Srivastava, A.K.; Cui, T.; Wang, Q.-E.; Wani, A.A. Differential DNA lesion formation and repair in heterochromatin and euchromatin. Carcinogenesis 2016, 37, 129–138. [Google Scholar] [CrossRef]
- Fan, W.; Luo, J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylate H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; di Fagagna, F.D.A. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Di Micco, R.; Fagagna, F.D.D. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-strand breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Tyler, J.K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell. Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Neste, L.V.; Easwaran, H.; et al. Oxidative Damage targets complexes containing DNA Methyltransferases, SIRT1 and Polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; Park, J.K.; Burneskis, J.M.; Barrett, C.; Horikawa. Evolutionarilly conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Bindu, S.; Sharp, W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. SIRT3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, R.P.; Kim, S.-J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.-H.; Jiang, H.; Kim, H.-S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. SIRT3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Deng, C.X.; Finley, H.S.; Kim, H.-S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging 2013, 5, 835–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.-B.; Jin, W.; Huang, H.-H.; Chen, X. SIRT5 desunccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef]
- Wilking, M.; Singh, C.K.; Nihal, M.; Ndiaye, M.A.; Ahmad, N. Sirtuin deacetylases: A new target for melanoma management. Cell Cycle 2014, 13, 2821–2826. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The critical role of the class II histone deacetylase SIRT1 in cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef]
- Ong, A.L.C.; Ramasamy, T.S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 2018, 43, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Ruscica, V.; Restelli, M.; Fontanella, E.; Buscemi, G.; Zannini, L. CCAR2/DBC1 is required for Chk2-dependent KAP1 phosphorylation and repair of DNA damage. Oncotarget 2015, 6, 17817–17831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magni, M.; Buscemi, G.; Maita, L.; Peng, L.; Chan, S.Y.; Montecucco, A.; Delia, D.; Zannini, L. TSPYL2 is a novel regulator of SIRT1 and p300 activity in response to DNA damage. Cell Death Differ. 2019, 26, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Buscemi, G.; Zannini, L. Cell cycle and apoptosis regulator 2 at the interface between DNA damage response and cell physiology. Mut. Res. Rev. 2018, 776, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Batta, K.; Das, C.; Gadad, S.; Shandilya, J.; Kundu, T.K. Reversible acetylation of nonhistone proteins: Role in cellular function and disease. Subcell. Biochem. 2007, 41, 193–212. [Google Scholar] [PubMed]
- Oberdoerffer, P.; Sinclair, D.A. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. 2007, 8, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Zhong, J.; Ji, L.; Chen, H.; Li, X.; Zhang, J.; Wang, X.; Wu, W.; Xu, Y.; Huang, F.; Cai, W.; et al. Acetylation of hMOF modulates H4K16ac to regulate DNA repair genes in response to oxidative stress. Int. J. Biol. Sci. 2017, 13, 923–934. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Erdjument-Bromage, H.; Tempst, P.; Serrano, L.; Reinverg, D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 2007, 450, 440–444. [Google Scholar] [CrossRef]
- Rifai, K.; Judes, G.; Idrissou, M.; Daures, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 2018, 9, 30661–30678. [Google Scholar] [CrossRef]
- Hida, Y.; Kubo, Y.; Murao, K.; Arase, S. Strong expression of a longevity-related protein, SIRT1, in Bowen’s disease. Arch. Dermatol. Res. 2007, 299, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.-S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Jiao, L.; Wang, Y.; Yu, Y.; Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 2018, 9, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Meliso, F.M.; Micali, D.; Silva, C.T.; Sabedot, T.S.; Coetzee, S.G.; Koch, A.; Fahlbusch, F.B.; Noushmehr, H.; Schneider-Stock, R.; Jasiulionis, M.G. SIRT1 regulates Mxd1 during malignant melanoma progression. Oncotarget 2017, 8, 114540–114553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.-S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef] [PubMed]
- Stunkel, W.; Peh, B.K.; Tan, Y.C.; Nayagam, V.M.; Wang, X.; Salto-Tellez, M.; Ni, B.; Entezeroth, M.; Wood, J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007, 2, 1360–1368. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.-H.; Min, B.-H.; Lee, K.-M.; Cho, M.-H.; Park, G.; Lee, K.-H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double Strand Breaks can initiate gene silencing and SIR1-dependent onset of DNA Methylation in an exogenous promoter CpG Island. PLoS Genet. 2008, 4, 1–16. [Google Scholar]
- O’Hagan, H.M. Chromatin modifications during Repair of Environmental Exposure-Induced DNA damage: A potential mechanism for stable epigenetic alterations. Environ. Mol. Mutagenesis 2014, 55, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Boham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O’Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ura, K.; Araki, M.; Saeki, H.; Masutani, C.; Ito, T.; Iwai, S.; Mizukoshi, T.; Kaneda, Y.; Hanaoka, F. ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J. 2001, 20, 2004–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketogenic Diet Modulates NAD+-dependent enzymes and reduces DNA damage in hippocampus. Front. Cell. Neurosci. 2018, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in healthy and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Horikoshi, N.; Singh, M.; Gupta, A.; Misra, H.S.; Albuquerque, K.; Hunt, C.R.; Pandita, T.K. Chromatin modifications and the DNA damage response to ionizing radiation. Front. Oncol. 2012, 2, 214. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.-K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. DNA damage-induced alterations in chromatin contribute to genomic integrity and age-related changes in gene expression. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef]
- Grustein, M. Molecular model for telomeric heterochromatin in yeast. Curr. Opin. Cell Biol. 1997, 9, 383–387. [Google Scholar] [CrossRef]
- Mills, K.D.; Sinclair, D.A.; Guarente, L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 1999, 97, 609–620. [Google Scholar] [CrossRef]
- Deng, C.X. SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar] [CrossRef]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-based combinatorial resveratrol and pterostilbene alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including SIRT1-dependent γH2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer 2015, 15, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Karl-Heinz, K.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to MYC cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, G.; Khoronenkova, S.V.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. ARF induction in response to DNA strand breaks is regulated by PARP1. Nucleic Acids Res. 2014, 42, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Matheu, A.; Maraver, A.; Serrano, M. The Arf/p53 pathway in cancer and aging. Cancer Res. 2008, 68, 6031–6034. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SIRT1 regulates apoptotic response to DNA damage. Nat. Cell. Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The deacetylase Sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog. 2015, 11, e1005181. [Google Scholar] [CrossRef]
- Wong, S.; Weber, J.D. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem. J. 2007, 407, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef]
- Hughes, K.J.; Meares, G.P.; Hansen, P.A.; Corbett, J.A. FoxO1 and SIRT1 regulates β-cell responses to nitric oxide. J. Biol. Chem. 2011, 286, 8338–8348. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakou, M.E.; Partridge, L. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell Biol. 2004, 14, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins in Aging and Disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, A.; Chen, C.; da Costa, S.V.; Sanchez, M.; Sokol, K.; Nussenzweig, M.C.; Li, G.C. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Nussenzweig, A.; Kurimasa, A.; Soares, V.C.; Li, X.; Cordon-Cardo, C.; Li, W.-H.; Cheong, N.; Nussenzweig, M.; Illiakis, G.; et al. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med. 1997, 186, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589–598. [Google Scholar] [CrossRef]
- Roth, M.; Wang, Z.; Chen, W.Y. SIRT1 and LSD1 competitively regulates Ku70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 2016, 7, 50195–50214. [Google Scholar] [CrossRef]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sayers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Li, L.; Wang, G.; Zhang, W.; Xu, J.; Liang, A. Ku70 inhibition impairs both Non-Homologous End Joining and Homologous Recombination DNA damage repair through SHP-1 induced dephosphorylation of SIRT1 in adult T-cell leukemia-lymphoma cells. Cell. Physiol. Biochem. 2018, 49, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Parry, P.; Wei, Y.; Evans, G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer 1994, 11, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Borkhardt, A.; Repp, R.; Haas, O.A.; Leis, T.; Harbott, J.; Kreuder, J.; Hammermann, J.; Henn, T.; Lampert, F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11) (q13;q23). Oncogene 1997, 14, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Kobayashi, S.; Bronk, S.F.; Bronk, S.F.; Werneburg, N.W.; Malhi, H.; Gores, G.J. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 2007, 282, 27141–27154. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [Green Version]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Yuan, Z.; Seto, E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle 2007, 6, 2869–2871. [Google Scholar] [CrossRef]
- Kahyo, T.; Mostoslavsky, R.; Goto, M.; Setou, M. Sirtiun-mediated deacetylation pathway stabilizes Werner syndrome protein. FEBS Lett. 2008, 582, 2479–2483. [Google Scholar] [CrossRef]
- Opresko, P.L.; Chen, W.H.; Kobbe, C.V.; Harrigan, J.A.; Bohr, V.A. Werner syndrome and the function of the Werner protein: What they can teach us about the molecular aging process. Carcinogenesis 2003, 24, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Comai, L.; Li, B. The Werner syndrome protein at the crossroads of DNA repair and apoptosis. Mech. Ageing Dev. 2004, 125, 521–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Yan, J.; Pei, H.; Liu, T.; Ann, D.K.; Lou, Z. KAP1 deacetylation promotes by SIRT1 promotes Non-Homologous End-Joining Repair. PLoS ONE 2015, 10, e0123935. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Carter, K.M.; Bautista, R.; Bautista, R.-M.; He, D.; Wang, C.; D’Orazio, J.A. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J. Biol. Chem. 2018, 293, 19025–19037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, T.; Wiederhold, L.R.; Roy, G.; Roy, R.; Jaiswal, A.; Bhakat, K.K.; Mitra, S.; Hazra, T.K. Mammalian DNA base excision repair proteins: Their interactions and role in repair of oxidative DNA damage. Toxicology 2003, 193, 43–65. [Google Scholar] [CrossRef]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.-B.; Kim, C.-S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Presgué, L.; Vaquero, A. The dual role of Sirtuin in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef]
- Martinez-Pastor, B.; Mostoslavsky, R. Sirtuins, metabolismo, and cancer. Front. Pharm. 2012, 3, 1–7. [Google Scholar] [CrossRef]
- Rajendran, R.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Sirtuins: Molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J. Biomed. Biotechnol. 2011, 2011, 368276. [Google Scholar] [CrossRef]


{kind=link}
| Role of SIRT1 | Effects | Cell or Tissue Type | Reference |
|---|---|---|---|
| Protection against DNA damage | Absence of 6-4PP and Pt-GG in heterochromatin associated with SIRT1 | Human fibroblasts | [20] |
| Decreased levels of 8-OHdG after the increase in SIRT1 activity and mRNA level | Rat hippocampus | [64] | |
| Regulation of genomic stability and transcriptional changes | Relocalization of Sir2/3/4 and de-repression of epigenetically silencing genes in response to DNA damage | Yeast | [69] |
| Relocalization of SIRT1 to DSBs followed by transcriptional changes | Mammalian stem cells | [67] | |
| Recruitment of key epigenetic proteins (DNMTs, EZH2) to DNA damage site | Normal and cancer cells | [28,55,60,62,71] | |
| Deacetylation of HAT (hMOF) and E2F1 leading to downregulation of genes involved in DNA repair and genomic stability | Cancer cells | [48,74] | |
| Recruitment of DNA repair proteins contributing with viral activity, including gene transcription | Keratinocytes containing HPV episomes | [78] | |
| Modulation of DDR and DNA repair | Deacetylation of p53 interfering in cell death | Normal and cancer cells | reviewed in [41] |
| Deacetylation of γH2AX, Rad51, BRCA1 and NBS1, regulating the foci formation | MEFs | [52] | |
| Interaction with Ku70 protein belonging to NHEJ | Cancer cells | [88,89,90,91,92] | |
| Deacetylation of FOXO family favoring the expression of target repair genes, cell cycle arrest and resistance to oxidative stress | Normal cells | [81,82,83,84] | |
| Regulation of many important proteins to DDR and DNA repair, including ATM, NBS1, WRN, KAP1, XPA, MSH2, MSH6, APEX1 | Normal and cancer cells | [11,97,99,100,101,102,103,104,106] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. https://doi.org/10.3390/ijms20133153
Alves-Fernandes DK, Jasiulionis MG. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. International Journal of Molecular Sciences. 2019; 20(13):3153. https://doi.org/10.3390/ijms20133153
Chicago/Turabian StyleAlves-Fernandes, Débora Kristina, and Miriam Galvonas Jasiulionis. 2019. "The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer" International Journal of Molecular Sciences 20, no. 13: 3153. https://doi.org/10.3390/ijms20133153
APA StyleAlves-Fernandes, D. K., & Jasiulionis, M. G. (2019). The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. International Journal of Molecular Sciences, 20(13), 3153. https://doi.org/10.3390/ijms20133153