Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy


Abstract
:1. Introduction
2. Tumor Milieu as a Critical Immune Mediator of RT
2.1. RT as A Frontline Anticancer Therapy
2.2. Immunogenic Cell Death in Irradiated TME
2.3. Innate and Adaptive Immune Response Activation
2.4. IR-Induced T Lymphocytes Activation
2.5. IR-Induced Tumor Infiltration of Immune Cells
2.6. Systemic Reaction of Irradiated TME
3. Immunosuppressive TME as a Side Effect of RT
3.1. Immunostimulation Processes Activate Radioresistance
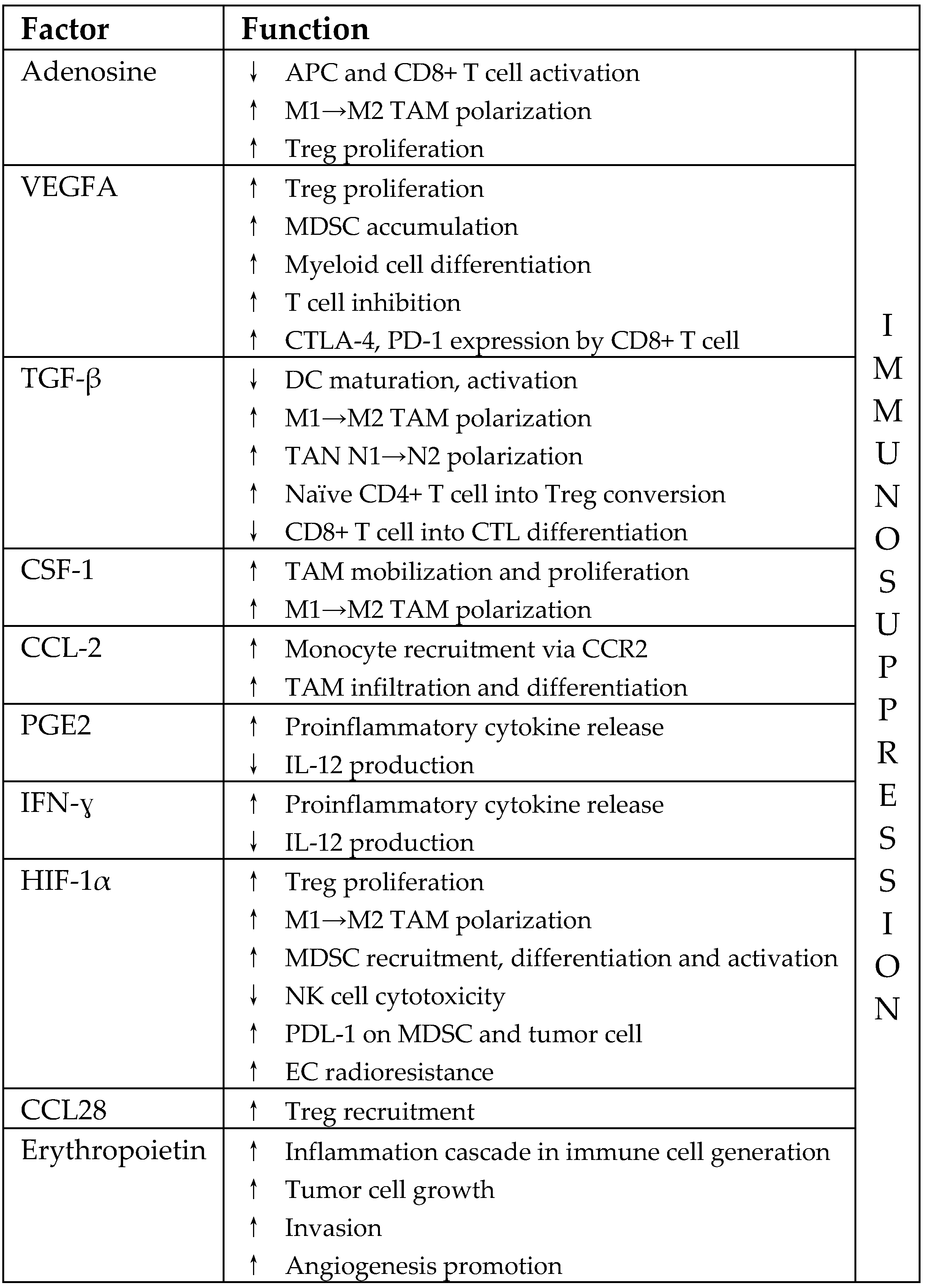
3.2. Immunosuppressive Pathways in Irradiated TME
3.3. IR-Induced Immunosuppressive Immune Cells
3.4. CAFs in Irradiated TME
4. Tumor Vasculature and RT
4.1. IR-Induced EC Dysfunctions
4.2. Pro-Survival Processes in the TME after EC Irradiation
4.3. Vascular Remodeling in Irradiated TME
4.4. IR-Induced Vasculogenesis
4.5. Hypoxia in Irradiated TME
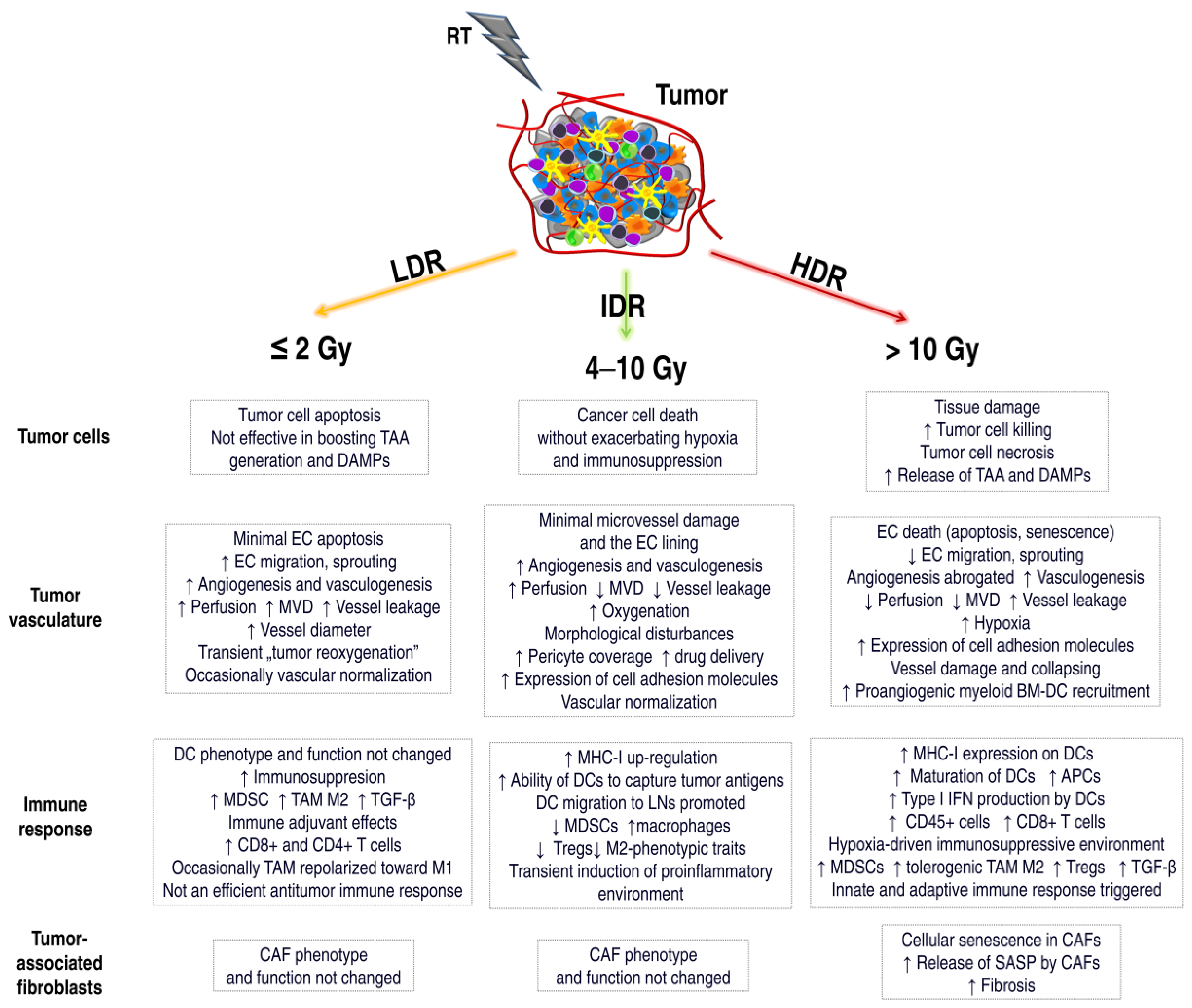
5. Different Responses of the TME after Various Doses of RT
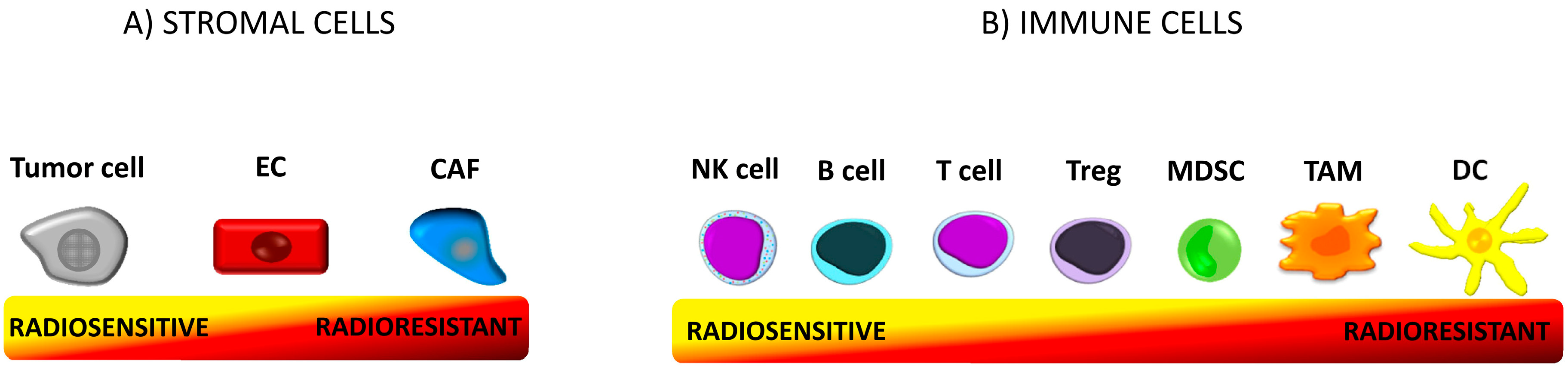
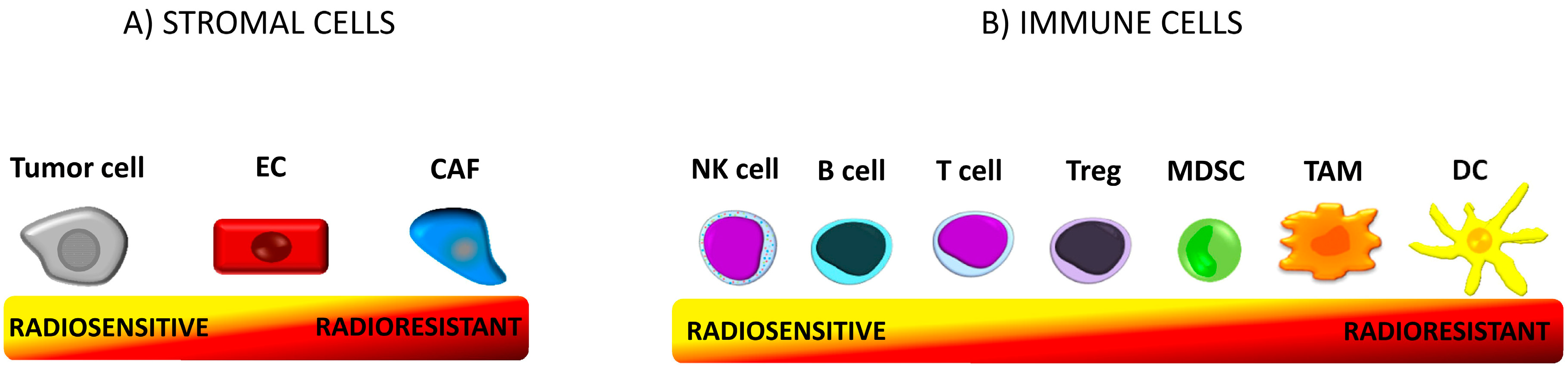
5.1. Radioresistance of Immune Cells
5.2. Immune Response in TME vs. Low Dose of RT
5.3. Immune Response in TME vs. High Doses of RT
5.4. EC Response vs. Doses of RT
5.5. Vascular Response in TME vs. Low Dose of RT
5.6. Vascular Response in the TME vs. High Dose of RT
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate immunity, inflammation and tumour progression: Double-edged swords. J. Intern. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H. Remodeling the Irradiated Tumor Microenvironment: The Fifth R of Radiobiology? In Increasing the Therapeutic Ratio of Radiotherapy. Cancer Drug Discovery and Development; Tofilon, P., Camphausen, K., Eds.; Humana Press: Cham, Switzerland, 2017; pp. 135–149. [Google Scholar]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Shekarian, T.; Valsesia-Wittmann, S.; Caux, C.; Marabelle, A. Paradigm shift in oncology: Targeting the immune system rather than cancer cells. Mutagenesis 2015, 30, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Guo, G. Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Tugues, S.; Ducimetiere, L.; Friebel, E.; Becher, B. Innate lymphoid cells as regulators of the tumor microenvironment. Semin. Immunol 2019. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the Tumor Microenvironment and Tumor-Stroma Interaction by Non-invasive Preclinical Imaging. Front. Oncol. 2017, 7, 3. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Kleibeuker, E.A.; Griffioen, A.W.; Verheul, H.M.; Slotman, B.J.; Thijssen, V.L. Combining angiogenesis inhibition and radiotherapy: A double-edged sword. Drug Resist. Updates 2012, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viallard, C.; LarriveÂe, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- El Alaoui-Lasmaili, K.; Faivre, B. Antiangiogenic therapy: Markers of response, “normalization” and resistance. Crit. Rev. Oncol. Hematol. 2018, 128, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, J. Present and future of cancer immunotherapy: A tumor microenvironmental perspective. Oncol. Lett. 2018, 16, 4105–4113. [Google Scholar] [CrossRef] [PubMed]
- Szala, S.; Jarosz-Biej, M.; Cichoń, T.; Smolarczyk, S. Polarization of tumor milieu: Therapeutic implications. In Cancer Immunology: Translational Medicine from Bench to Bedside; Part II: Cancer Immunotherapy; Rezaei, N., Ed.; Springer: Heidelberg, Germany, 2015; pp. 401–408. [Google Scholar]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.Z.; Jin, M.Z.; Dai, J.X.; Sun, W.; Feng, J.H.; Jin, W.L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; DeClerck, Y.A. Tumor microenvironment complexity: Emerging roles in cancer therapy. Cancer Res. 2012, 72, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage Polarization: Anti-Cancer Strategies to Target Tumor-Associated Macrophage in Breast Cancer. J. Cell. Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Vatner, R.E.; Formenti, S.C. Myeloid-derived cells in tumors: Effects of radiation. Semin. Radiat. Oncol. 2015, 25, 18–27. [Google Scholar] [CrossRef]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019, 101174. [Google Scholar] [CrossRef]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Pitt, J.M.; Li, Q.; Yang, H. The renaissance of anti-neoplastic immunity from tumor cell demise. Immunol. Rev. 2017, 280, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Jankowska, P.; Hingorani, M. Molecular biology for the radiation oncologist: The 5Rs of radiobiology meet the hallmarks of cancer. Clin. Oncol. 2007, 19, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Good, J.S.; Harrington, K.J. The hallmarks of cancer and the radiation oncologist: Updating the 5Rs of radiobiology. Clin. Oncol. 2013, 25, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hekim, N.; Cetin, Z.; Nikitaki, Z.; Cort, A.; Saygili, E.I. Radiation triggering immune response and inflammation. Cancer Lett. 2015, 368, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Jin, S.; Zhang, A.; Zhang, B.; Shi, X.; Wang, J.; Zhao, Y. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat. Res. 2010, 173, 148–157. [Google Scholar] [CrossRef]
- Persa, E.; Balogh, A.; Sáfrány, G.; Lumniczky, K. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. 2015, 368, 252–261. [Google Scholar] [CrossRef]
- Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef]
- Herrera, F.G.; Bourhis, J.; Coukos, G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J. Clin. 2017, 67, 65–85. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef]
- Falcke, S.E.; Rühle, P.F.; Deloch, L.; Fietkau, R.; Frey, B.; Gaipl, U.S. Clinically Relevant Radiation Exposure Differentially Impacts Forms of Cell Death in Human Cells of the Innate and Adaptive Immune System. Int. J. Mol. Sci. 2018, 19, 3574. [Google Scholar] [CrossRef] [PubMed]
- Ozpiskin, O.M.; Zhang, L.; Li, J.J. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics 2019, 9, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Chandna, S. Radiation-induced inflammatory cascade and its reverberating crosstalks as potential cause of post-radiotherapy second malignancies. Cancer Metastasis Rev. 2017, 36, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Frey, B.; Fietkau, R.; Gaipl, U.S. Immune-modulating properties of ionizing radiation: Rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol. Immunother. 2016, 65, 779–786. [Google Scholar] [CrossRef]
- Chajon, E.; Castelli, J.; Marsiglia, H.; De Crevoisier, R. The synergistic effect of radiotherapy and immunotherapy: A promising but not simple partnership. Crit. Rev. Oncol. Hematol. 2017, 111, 124–132. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef]
- Lauber, K.; Ernst, A.; Orth, M.; Herrmann, M.; Belka, C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front. Oncol. 2012, 2, 116. [Google Scholar] [CrossRef] [Green Version]
- Deloch, L.; Derer, A.; Hartmann, J.; Frey, B.; Fietkau, R.; Gaipl, U.S. Modern Radiotherapy Concepts and the Impact of Radiation on Immune Activation. Front. Oncol. 2016, 6, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Formenti, S.C. Role of T lymphocytes in tumor response to radiotherapy. Front. Oncol. 2012, 2, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locy, H.; de Mey, S.; de Mey, W.; De Ridder, M.; Thielemans, K.; Maenhout, S.K. Immunomodulation of the Tumor Microenvironment: Turn Foe Into Friend. Front. Immunol. 2018, 9, 2909. [Google Scholar] [CrossRef] [PubMed]
- Wirsdörfer, F.; de Leve, S.; Jendrossek, V. Combining Radiotherapy and Immunotherapy in Lung Cancer: Can We Expect Limitations Due to Altered Normal Tissue Toxicity? Int. J. Mol. Sci. 2018, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Rückert, M.; Deloch, L.; Fietkau, R.; Frey, B.; Hecht, M.; Gaipl, U.S. Immune modulatory effects of radiotherapy as basis for well-reasoned radioimmunotherapies. Strahlenther. Onkol. 2018, 194, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsou, P.G.; Zaman, K.; Martin Lluesma, S.; Cagnon, L.; Kandalaft, L.; Vozenin, M.C. Emerging Opportunities of Radiotherapy Combined With Immunotherapy in the Era of Breast Cancer Heterogeneity. Front. Oncol. 2018, 8, 609. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Hammerich, L.; Bhardwaj, N.; Kohrt, H.E.; Brody, J.D. In situ vaccination for the treatment of cancer. Immunotherapy 2016, 8, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Pilones, K.A.; García-Martínez, E.; Rudqvist, N.P.; Formenti, S.C.; Demaria, S. Barriers to Radiation-Induced In Situ Tumor Vaccination. Front. Immunol. 2017, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.M.; Flynn, N.J.; Raben, A.; Romak, L.; Yu, Y.; Dicker, A.P.; Mourtada, F.; Sims-Mourtada, J. The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 2018, 11, 1179064418761639. [Google Scholar] [CrossRef] [PubMed]
- Ebner, D.K.; Tinganelli, W.; Helm, A.; Bisio, A.; Yamada, S.; Kamada, T.; Shimokawa, T.; Durante, M. The Immunoregulatory Potential of Particle Radiation in Cancer Therapy. Front. Immunol. 2017, 8, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Frey, B.; Rubner, Y.; Wunderlich, R.; Weiss, E.M.; Pockley, A.G.; Fietkau, R.; Gaipl, U.S. Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation—implications for cancer therapies. Curr. Med. Chem. 2012, 19, 1751–1764. [Google Scholar] [CrossRef]
- Bockel, S.; Durand, B.; Deutsch, E. Combining radiation therapy and cancer immune therapies: From preclinical findings to clinical applications. Cancer Radiother. 2018, 22, 567–580. [Google Scholar] [CrossRef]
- Park, B.; Yee, C.; Lee, K.M. The effect of radiation on the immune response to cancers. Int. J. Mol. Sci. 2014, 15, 927–943. [Google Scholar] [CrossRef]
- Frey, B.; Rubner, Y.; Kulzer, L.; Werthmöller, N.; Weiss, E.M.; Fietkau, R.; Gaipl, U.S. Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol. Immunother. 2014, 63, 29–36. [Google Scholar] [CrossRef]
- Gandhi, S.J.; Minn, A.J.; Vonderheide, R.H.; Wherry, E.J.; Hahn, S.M.; Maity, A. Awakening the immune system with radiation: Optimal dose and fractionation. Cancer Lett. 2015, 368, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Formenti, S.C. Is tumor (R)ejection by the immune system the “5th R” of radiobiology? Oncoimmunology 2014, 3, e28133. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Na.t Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Bose, D. cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response. Int. J. Mol. Sci. 2017, 18, 2456. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, O.; Nowis, D. STING Signaling in Cancer Cells: Important or Not? Arch. Immunol. Ther. Exp. 2018, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar]
- Bernier, J. Immuno-oncology: Allying forces of radio- and immuno-therapy to enhance cancer cell killing. Crit. Rev. Oncol. Hematol. 2016, 108, 97–108. [Google Scholar] [CrossRef]
- Menon, H.; Ramapriyan, R.; Cushman, T.R.; Verma, V.; Kim, H.H.; Schoenhals, J.E.; Atalar, C.; Selek, U.; Chun, S.G.; Chang, J.Y.; et al. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front. Immunol. 2019, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Hanna, G.G.; Coyle, V.M.; Prise, K.M. Immune modulation in advanced radiotherapies: Targeting out-of-field effects. Cancer Lett. 2015, 368, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhou, T.; Liu, W.; Zuo, L. Molecular mechanism of bystander effects and related abscopal/cohort effects in cancer therapy. Oncotarget 2018, 9, 18637–18647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander effects and radiotherapy. Rep. Pract. Oncol. Radiother. 2014, 20, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Klammer, H.; Mladenov, E.; Li, F.; Iliakis, G. Bystander effects as manifestation of intercellular communication of DNA damage and of the cellular oxidative status. Cancer Lett. 2015, 356, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Chakraborty, A. Radiation-induced bystander phenomenon: Insight and implications in radiotherapy. Int. J. Radiat. Biol. 2019, 95, 243–263. [Google Scholar] [CrossRef] [PubMed]
- Meziani, L.; Deutsch, E.; Mondini, M. Macrophages in radiation injury: A new therapeutic target. Oncoimmunology 2018, 7, e1494488. [Google Scholar] [CrossRef]
- Mole, R.H. Whole body irradiation; radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Deplanque, G.; Shabafrouz, K.; Obeid, M. Can local radiotherapy and IL-12 synergise to overcome the immunosuppressive tumor microenvironment and allow “in situ tumor vaccination”? Cancer Immunol. Immunother. 2017, 66, 833–840. [Google Scholar] [CrossRef]
- Hu, Z.I.; McArthur, H.L.; Ho, A.Y. The Abscopal Effect of Radiation Therapy: What Is It and How Can We Use It in Breast Cancer? Curr. Breast Cancer Rep. 2017, 9, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Ouyang, Z. Following the Preclinical Data: Leveraging the Abscopal Effect More Efficaciously. Front. Oncol. 2017, 7, 66. [Google Scholar] [CrossRef]
- Sheen, M.R.; Fiering, S. In situ vaccination: Harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1524. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Hudson, A.L.; Back, M.; Eade, T.; Diakos, C.I. Radiation, inflammation and the immune response in cancer. Mamm. Genome 2018, 29, 843–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, J.M.; Shinohara, E.; Summers, J.B.; Niermann, K.J.; Morimoto, A.; Brousal, J. The controversial abscopal effect. Cancer Treat. Rev. 2005, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.G.; Yuan, H.; Deng, W.; Li, J.; Huang, Y.; Kim, B.Y.S.; Story, M.D.; Jiang, W. The Reciprocity between Radiotherapy and Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, E.; Vanpouille-Box, C.; Bornstein, S.; Yamazaki, T.; Demaria, S.; Galluzzi, L. Immune recognition of irradiated cancer cells. Immunol. Rev. 2017, 280, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Horn, L.A.; Ciavattone, N.G. Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy. Front. Oncol. 2019, 9, 215. [Google Scholar] [CrossRef]
- Wang, S.J.; Haffty, B. Radiotherapy as a New Player in Immuno-Oncology. Cancers 2018, 10, 515. [Google Scholar] [CrossRef]
- Jeong, H.; Bok, S.; Hong, B.J.; Choi, H.S.; Ahn, G.O. Radiation-induced immune responses: Mechanisms and therapeutic perspectives. Blood Res. 2016, 51, 157–163. [Google Scholar] [CrossRef]
- Hoves, S.; Ooi, C.H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Buranych, A.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. The role of tumor-associated macrophages in tumor vascularization. Vasc. Cell 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Shiao, S.L. The role of macrophage phenotype in regulating the response to radiation therapy. Transl. Res. 2018, 191, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; McBride, W.H. T lymphocytes and normal tissue responses to radiation. Front. Oncol. 2012, 2, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.W.; Bump, E.A.; Kim, J.S.; Janigro, D.; Mayberg, M.R. Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat. Res. 2001, 156, 232–240. [Google Scholar] [CrossRef]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161. [Google Scholar] [CrossRef]
- Venkatesulu, B.P.; Mahadevan, L.S.; Aliru, M.L.; Yang, X.; Bodd, M.H.; Singh, P.K.; Yusuf, S.W.; Abe, J.I.; Krishnan, S. Radiation-Induced Endothelial Vascular Injury: A Review of Possible Mechanisms. JACC Basic Transl. Sci. 2018, 3, 563–572. [Google Scholar] [CrossRef]
- Jaillet, C.; Morelle, W.; Slomianny, M.C.; Paget, V.; Tarlet, G.; Buard, V.; Selbonne, S.; Caffin, F.; Rannou, E.; Martinez, P.; et al. Radiation-induced changes in the glycome of endothelial cells with functional consequences. Sci. Rep. 2017, 7, 5290. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Deng, L.; Lu, Y. Challenges and opportunities of using stereotactic body radiotherapy with anti-angiogenesis agents in tumor therapy. Chin. J. Cancer Res. 2018, 30, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell 2004, 5, 405–406. [Google Scholar] [CrossRef] [Green Version]
- Nguemgo Kouam, P.; Bühler, H.; Hero, T.; Adamietz, I.A. The increased adhesion of tumor cells to endothelial cells after irradiation can be reduced by FAK-inhibition. Radiat. Oncol. 2019, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Vasculogenesis: A crucial player in the resistance of solid tumours to radiotherapy. Br. J. Radiol. 2014, 87, 20130686. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zubiaurre, I.; Chalmers, A.J.; Hellevik, T. Radiation-Induced Transformation of Immunoregulatory Networks in the Tumor Stroma. Front. Immunol. 2018, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.O.; Brown, J.M. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: Role of bone marrow-derived myelomonocytic cells. Cancer Cell 2008, 13, 193–205. [Google Scholar] [CrossRef]
- Kozin, S.V.; Kamoun, W.S.; Huang, Y.; Dawson, M.R.; Jain, R.K.; Duda, D.G. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010, 70, 5679–5685. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef]
- Leith, J.T.; Mousa, S.A.; Hercbergs, A.; Lin, H.Y.; Davis, P.J. Radioresistance of cancer cells, integrin αvβ3 and thyroid hormone. Oncotarget 2018, 9, 37069–37075. [Google Scholar] [CrossRef]
- Hill, R.P.; Bristow, R.G.; Fyles, A.; Koritzinsky, M.; Milosevic, M.; Wouters, B.G. Hypoxia and Predicting Radiation Response. Semin. Radiat. Oncol. 2015, 25, 260–272. [Google Scholar] [CrossRef]
- Smolarczyk, R.; Cichoń, T.; Pilny, E.; Jarosz-Biej, M.; Poczkaj, A.; Kułach, N.; Szala, S. Combination of anti-vascular agent - DMXAA and HIF-1α inhibitor - digoxin inhibits the growth of melanoma tumors. Sci. Rep. 2018, 8, 7355. [Google Scholar] [CrossRef]
- Soukup, K.; Wang, X. Radiation meets immunotherapy - a perfect match in the era of combination therapy? Int. J. Radiat. Biol. 2015, 91, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Manda, K.; Glasow, A.; Paape, D.; Hildebrandt, G. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front. Oncol. 2012, 2, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubner, Y.; Wunderlich, R.; Rühle, P.F.; Kulzer, L.; Werthmöller, N.; Frey, B.; Weiss, E.M.; Keilholz, L.; Fietkau, R.; Gaipl, U.S. How does ionizing irradiation contribute to the induction of anti-tumor immunity? Front. Oncol. 2012, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Hellevik, T.; Martinez-Zubiaurre, I. Radiotherapy and the tumor stroma: The importance of dose and fractionation. Front. Oncol. 2014, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Beyranvand Nejad, E.; Welters, M.J.; Arens, R.; van der Burg, S.H. The importance of correctly timing cancer immunotherapy. Expert. Opin. Biol. Ther. 2017, 17, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS⁺/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; To, N.H.; Zadigue, P.; Kerbrat, S.; De La Taille, A.; Le Gouvello, S.; Belkacemi, Y. Ionizing radiation-induced cellular senescence promotes tissue fibrosis after radiotherapy. A review. Crit. Rev. Oncol. Hematol. 2018, 129, 13–26. [Google Scholar] [CrossRef]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Potiron, V.A.; Abderrahmani, R.; Clément-Colmou, K.; Marionneau-Lambot, S.; Oullier, T.; Paris, F.; Supiot, S. Improved functionality of the vasculature during conventionally fractionated radiation therapy of prostate cancer. PLoS ONE 2013, 8, e84076. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. https://doi.org/10.3390/ijms20133212
Jarosz-Biej M, Smolarczyk R, Cichoń T, Kułach N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. International Journal of Molecular Sciences. 2019; 20(13):3212. https://doi.org/10.3390/ijms20133212
Chicago/Turabian StyleJarosz-Biej, Magdalena, Ryszard Smolarczyk, Tomasz Cichoń, and Natalia Kułach. 2019. "Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy" International Journal of Molecular Sciences 20, no. 13: 3212. https://doi.org/10.3390/ijms20133212
APA StyleJarosz-Biej, M., Smolarczyk, R., Cichoń, T., & Kułach, N. (2019). Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. International Journal of Molecular Sciences, 20(13), 3212. https://doi.org/10.3390/ijms20133212