Deletion of OGG1 Results in a Differential Signature of Oxidized Purine Base Damage in mtDNA Regions

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
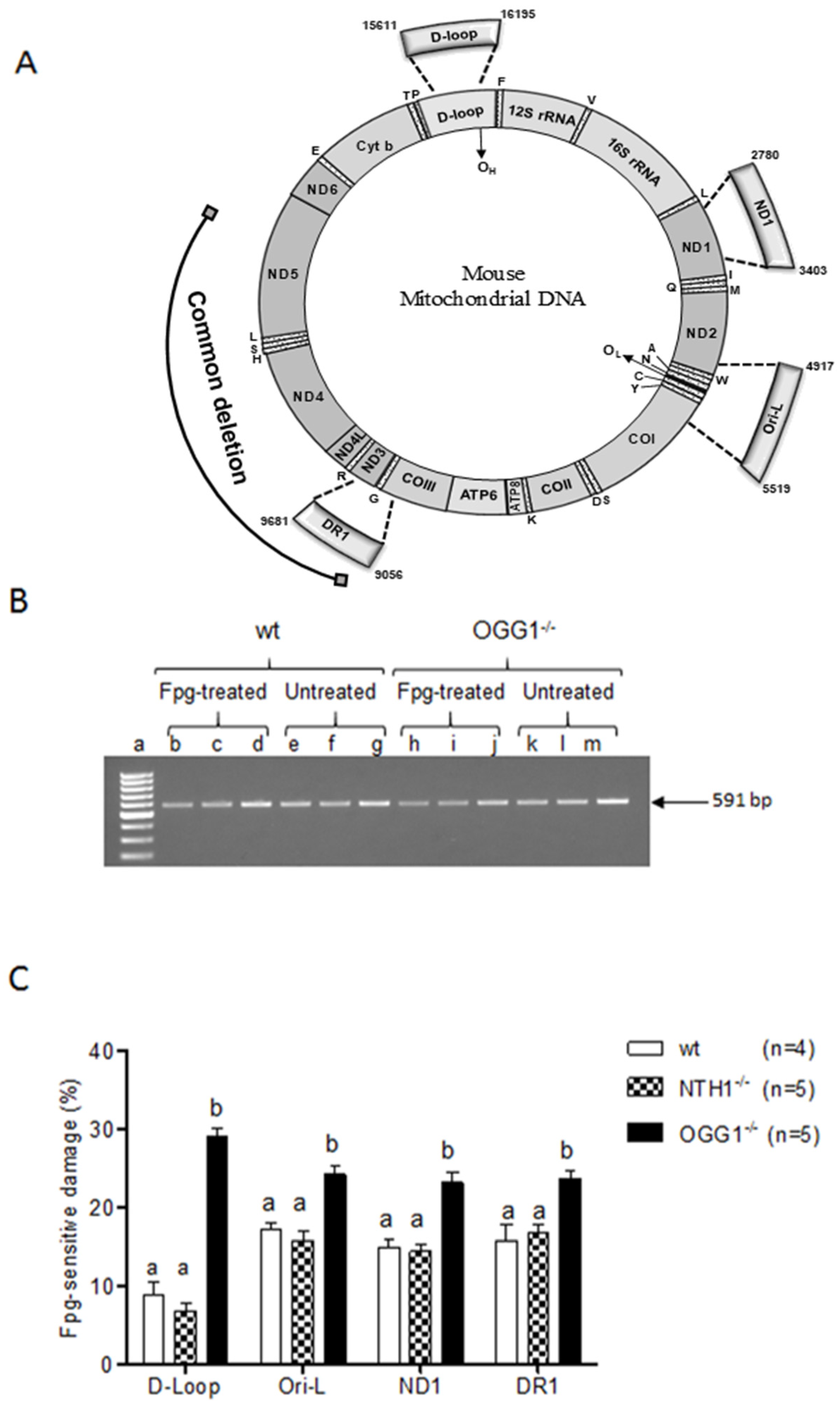
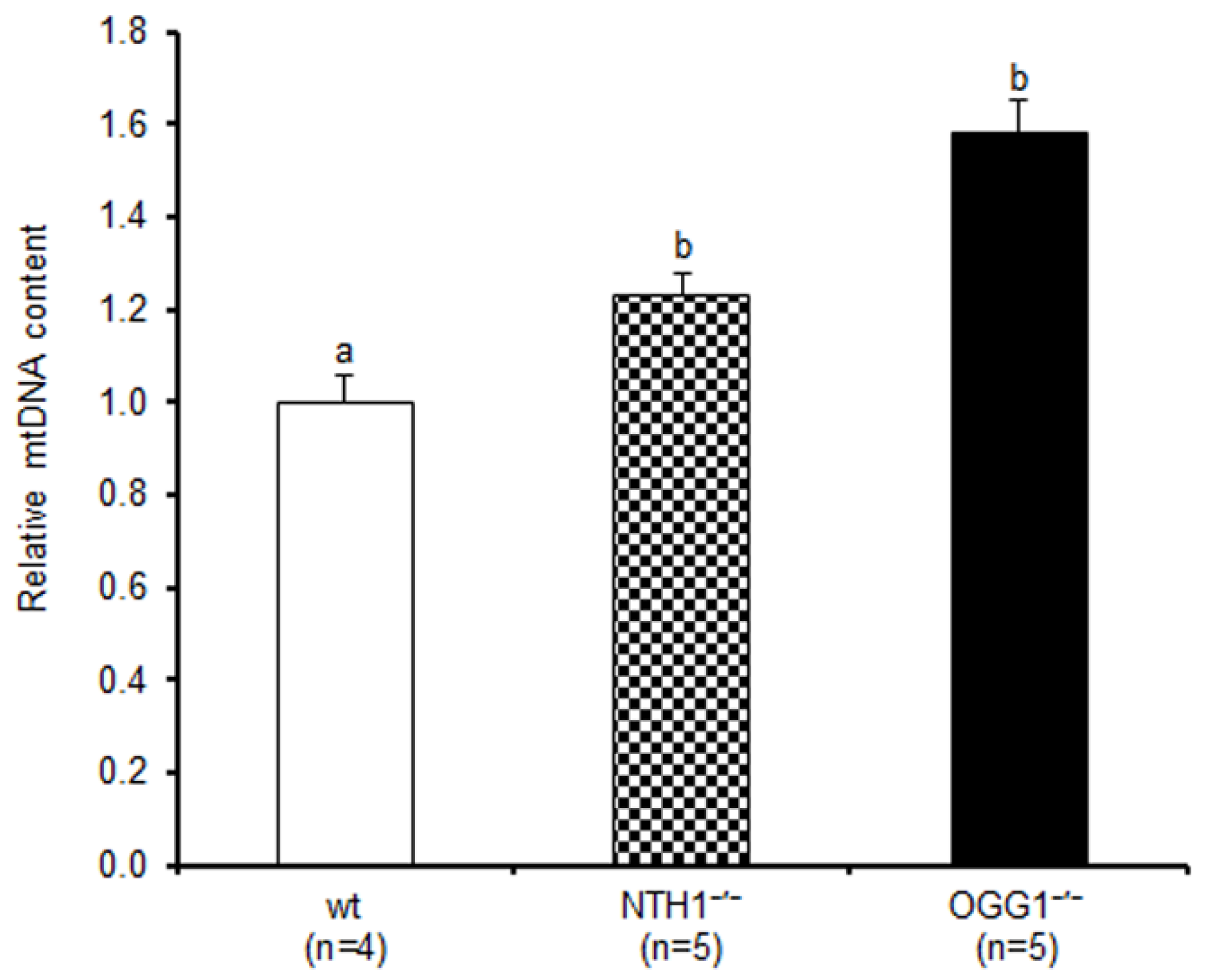
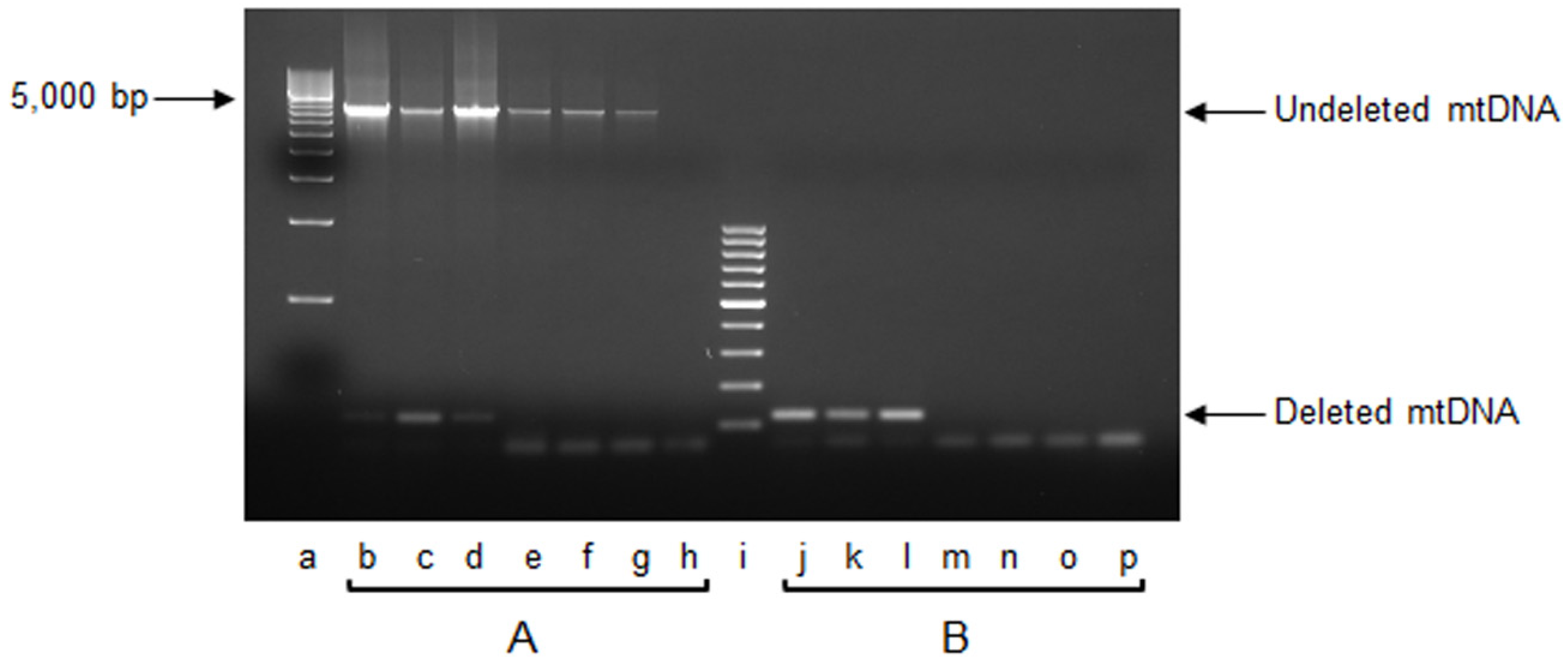
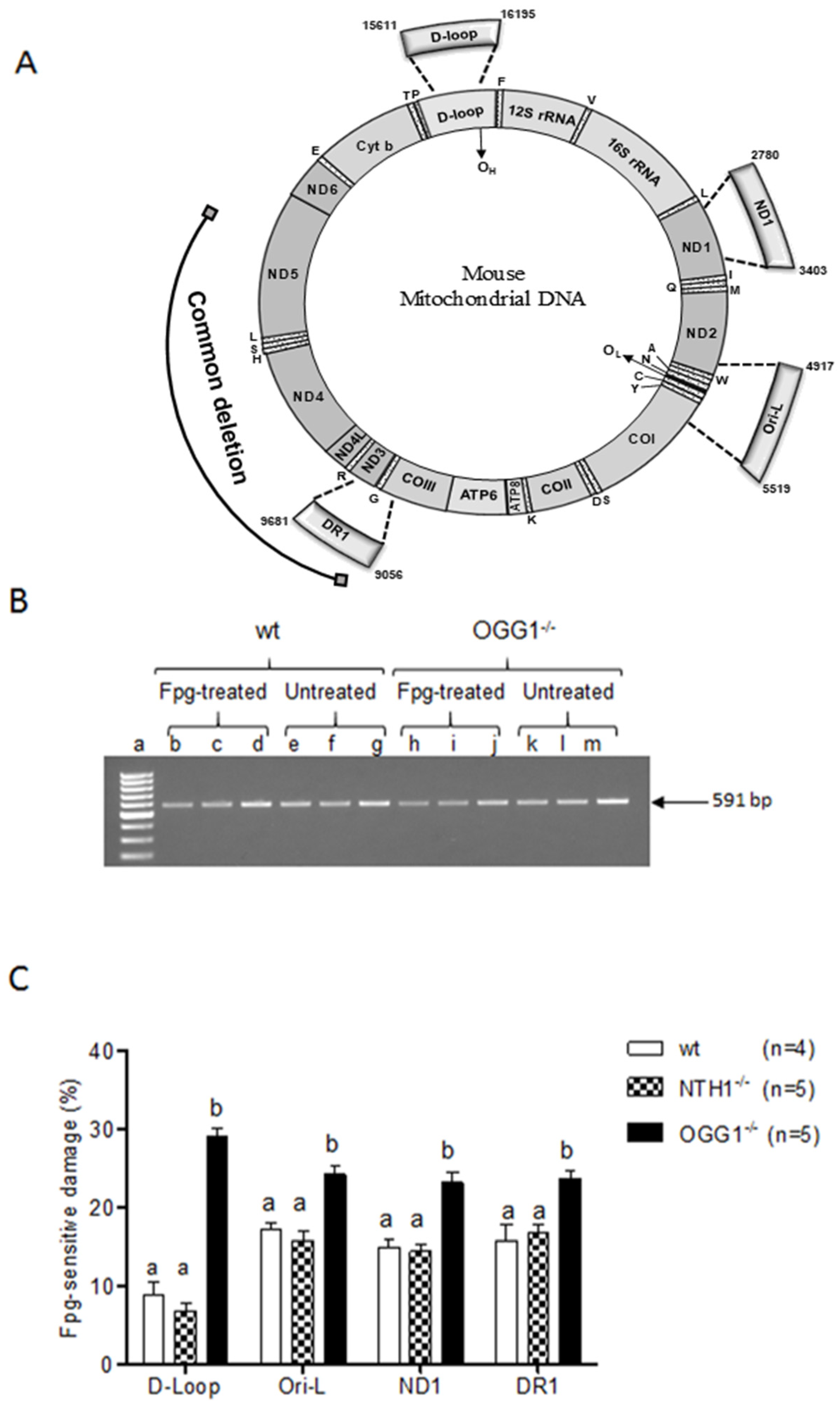
2.1. mtDNA Content and 3873-bp-Long Deletion
2.2. Analysis of Oxidized Purines
3. Discussion
3.1. Effects on mtDNA Content and Integrity
3.2. Oxidized Purine Damage at Specific mtDNA Regions
4. Materials and Methods
4.1. Mouse Samples
4.2. Isolation of Liver Genomic DNA
4.3. Measurement of mtDNA Content and Detection of mtDNA 3873-bp-Long Deletion
4.4. Modified Purines Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| OGG1 | 8-oxoG DNA glycosylase/AP lyase |
| mtDNA | mitochondrial DNA |
| ROS | reactive oxygen species |
| SSBs and DSBs | single- and double-strand breaks |
| BER | base excision repair |
| NTH1 | endonuclease III-like protein 1 |
| Fpg | formamidopyrimidine DNA glycosylase |
References
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, C.; Mayoral, P.; Quirós, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G.; Hernández-Camacho, J.D.; Fernández-Ayala, D.J.M.; Navas, P. Mitochondrial dysfunction in metabolism and ageing: Shared mechanisms and outcomes? Biogerontology 2018, 19, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Pesce, V.; Cormio, A.; Fracasso, F.; Vecchiet, J.; Felzani, G.; Lezza, A.M.; Cantatore, P.; Gadaleta, M.N. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic. Biol. Med. 2001, 30, 1223–1233. [Google Scholar] [CrossRef]
- Masuyama, M.; Iida, R.; Takatsuka, H.; Yasuda, T.; Matsuki, T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim. Biophys. Acta 2005, 1723, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.M.; Leeuwenburgh, C.; Lezza, A.M. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim. Biophys. Acta 2014, 1840, 2184–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.N.; Parsade, C.K.; Ajaz, S.; Crosby-Nwaobi, R.; Gnudi, L.; Czajka, A.; Sivaprasad, S. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res. Clin. Pract. 2015, 110, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Riezzo, G.; Lezza, A.M.S.; Clemente, C.; Pesce, V.; Orlando, A.; Chimienti, G.; Russo, F. Mitochondria and redox balance in coeliac disease: A case-control study. Eur. J. Clin. Investig. 2018, 48, e12877. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadaleta, M.N.; Rainaldi, G.; Lezza, A.M.; Milella, F.; Fracasso, F.; Cantatore, P. Mitochondrial DNA copy number and mitochondrial DNA deletion in adult and senescent rats. Mutat. Res. 1992, 275, 181–193. [Google Scholar] [CrossRef]
- Brossas, J.Y.; Barreau, E.; Courtois, Y.; Tréton, J. Multiple deletions in mitochondrial DNA are present in senescent mouse brain. Biochem. Biophys. Res. Commun. 1994, 202, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Sciacco, M.; Bonilla, E.; Schon, E.A.; DiMauro, S.; Moraes, C.T. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 1994, 3, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef]
- Leandro, G.S.; Sykora, P.; Bohr, V.A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. 2015, 776, 31–39. [Google Scholar] [CrossRef] [Green Version]
- de Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Karahalil, B.; de Souza-Pinto, N.C.; Parsons, J.L.; Elder, R.H.; Bohr, V.A. Compromised incision of oxidized pyrimidines in liver mitochondria of mice deficient in NTH1 and OGG1 glycosylases. J. Biol. Chem. 2003, 278, 33701–33707. [Google Scholar] [CrossRef]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, R.H.; Dianov, G.L. Repair of dihydrouracil supported by base excision repair in mNTH1 knock-out cell extracts. J. Biol. Chem. 2002, 277, 50487–50490. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sun, Y.; Hu, Y.J.; Yang, Y.; Yao, L.L.; Zhou, X.X.; Wang, H.; Zhang, R.; Huang, X.; Kong, W.J. Increased p66Shc in the inner ear of D-galactose-induced aging mice with accumulation of mitochondrial DNA 3873-bp deletion: p66Shc and mtDNA damage in the inner ear during aging. PLoS ONE 2012, 7, e50483. [Google Scholar] [CrossRef] [PubMed]
- de Souza-Pinto, N.C.; Hogue, B.A.; Bohr, V.A. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radic. Biol. Med. 2001, 30, 916–923. [Google Scholar] [CrossRef]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280, 40544–40551. [Google Scholar] [CrossRef]
- Stuart, J.A.; Bourque, B.M.; de Souza-Pinto, N.C.; Bohr, V.A. No evidence of mitochondrial respiratory dysfunction in OGG1-null mice deficient in removal of 8-oxodeoxyguanine from mitochondrial DNA. Free Radic. Biol. Med. 2005, 38, 737–745. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell. Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Milella, F.; Marra, M.; Pala, M.; Lezza, A.M.; Bonfigli, A.R.; Franceschi, C.; Cantatore, P.; Gadaleta, M.N. Variations at the H-strand replication origins of mitochondrial DNA and mitochondrial DNA content in the blood of type 2 diabetes patients. Biochim. Biophys. Acta 2009, 1787, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Chen, X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013, 77, 476–496. [Google Scholar] [CrossRef]
- Nissanka, N.; Bacman, S.R.; Plastini, M.J.; Moraes, C.T. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef] [PubMed]
- Pastukh, V.M.; Gorodnya, O.M.; Gillespie, M.N.; Ruchko, M.V. Regulation of mitochondrial genome replication by hypoxia: The role of DNA oxidation in D-loop region. Free Radic. Biol. Med. 2016, 96, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbisà, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Cantatore, P.; Daddabbo, L.; Fracasso, F.; Gadaleta, M.N. Identification by in Organello footprinting of protein contact sites and of single-stranded DNA sequences in the regulatory region of rat mitochondrial DNA. Protein binding sites and single-stranded DNA regions in isolated rat liver mitochondria. J. Biol. Chem. 1995, 270, 25020–25027. [Google Scholar] [CrossRef] [PubMed]
- Ghivizzani, S.C.; Madsen, C.S.; Nelen, M.R.; Ammini, C.V.; Hauswirth, W.W. In organelle footprint analysis of human mitochondrial DNA: Human mitochondrial transcription factor A interactions at the origin of replication. Mol. Cell. Biol. 1994, 14, 7717–7730. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.; Akie, T.E.; Sanosaka, M.; Craige, S.M.; Kant, S.; Keaney, J.F., Jr.; Cooper, M.P. Mitochondrial retrograde signaling connects respiratory capacity to thermogenic gene expression. Sci. Rep. 2017, 7, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 2017, 292, 20694–20706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial Retrograde Signaling in Mammals Is Mediated by the Transcriptional Cofactor GPS2 via Direct Mitochondria-to-Nucleus Translocation. Mol. Cell. 2018, 69, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, e51697. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, K.; Rachek, L.; You, P.; Rowe, A.D.; Wang, W.; Kuśnierczyk, A.; Kittelsen, L.; Bjoras, M.; Eide, L. 8-oxoguanine DNA glycosylase (Ogg1) controls hepatic gluconeogenesis. DNA Repair (Amst) 2018, 61, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prates Mori, M.; de Souza-Pinto, N.C. Role of mitochondrial dysfunction in the pathophysiology of DNA repair disorders. Cell Biol. Int. 2018, 42, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.M.; Leeuwenburgh, C.; Lezza, A.M.S. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS ONE 2013, 8, e74644. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| mtDNA Region | ||||
|---|---|---|---|---|
| Strain | d-loop | Ori-l | ND1 | DR1 |
| wt | 8.81 ± 1.64 a | 17.16 ± 0.94 b | 14.93 ± 1.12 ab | 15.73 ± 2.08 b |
| NTH1−/− | 6.79 ± 1.05 a | 15.77 ± 1.25 b | 14.49 ± 0.87 b | 16.77 ± 1.00 b |
| OGG1−/− | 29.10 ± 1.04 a | 24.27 ± 1.17 b | 23.20 ± 1.36 b | 23.79 ± 0.85 b |
| Primer Set | Forward Primer (5’–3’) | Reverse Primer (5’–3’) | (nps) | (nps) |
|---|---|---|---|---|
| mtDNA | AATCTACCATCCTCCGTGAAACC | GCCCGGAGCGAGAAGAG | 15,687–15,709 | 15,748–15,732 |
| β-actin | AGCCATGTACGTAGCCATCCA | TCTCCGGAGTCCATCACAATG | 499–519 | 579–559 |
| 3.8 Del | AGCCTTATAGAAGGTAAACGAAACC | ACGCGGTTTTGTTATTGTTACG | 9054–9078 | 13,050–13,029 |
| ND1 long | CGTCCCCATTCTAATCGCCA | AAGGCTACGGCAAATTCAAG | 2780–2799 | 3403–3384 |
| Ori-l long | ATAACCCTACCCCTAGCCCC | ACAAAAGCATGGGCAGTTACG | 4916–4935 | 5518–5498 |
| DR1 long | GCCTAATAGAAGGTAAACGAAACC | CTATTCCTGCTCAGGCTCCA | 9055–9078 | 9680–9656 |
| d-loop long | GTGTTATCTGACATACACCATACAG | TGGGAACTACTAGAATTGATCAGGA | 15,611–15,635 | 16,201–16,177 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chimienti, G.; Pesce, V.; Fracasso, F.; Russo, F.; de Souza-Pinto, N.C.; Bohr, V.A.; Lezza, A.M.S. Deletion of OGG1 Results in a Differential Signature of Oxidized Purine Base Damage in mtDNA Regions. Int. J. Mol. Sci. 2019, 20, 3302. https://doi.org/10.3390/ijms20133302
Chimienti G, Pesce V, Fracasso F, Russo F, de Souza-Pinto NC, Bohr VA, Lezza AMS. Deletion of OGG1 Results in a Differential Signature of Oxidized Purine Base Damage in mtDNA Regions. International Journal of Molecular Sciences. 2019; 20(13):3302. https://doi.org/10.3390/ijms20133302
Chicago/Turabian StyleChimienti, Guglielmina, Vito Pesce, Flavio Fracasso, Francesco Russo, Nadja Cristhina de Souza-Pinto, Vilhelm A. Bohr, and Angela Maria Serena Lezza. 2019. "Deletion of OGG1 Results in a Differential Signature of Oxidized Purine Base Damage in mtDNA Regions" International Journal of Molecular Sciences 20, no. 13: 3302. https://doi.org/10.3390/ijms20133302