Whole Genome Microarray Analysis of DUSP4-Deletion Reveals A Novel Role for MAP Kinase Phosphatase-2 (MKP-2) in Macrophage Gene Expression and Function

 ,
,
Abstract
:1. Introduction
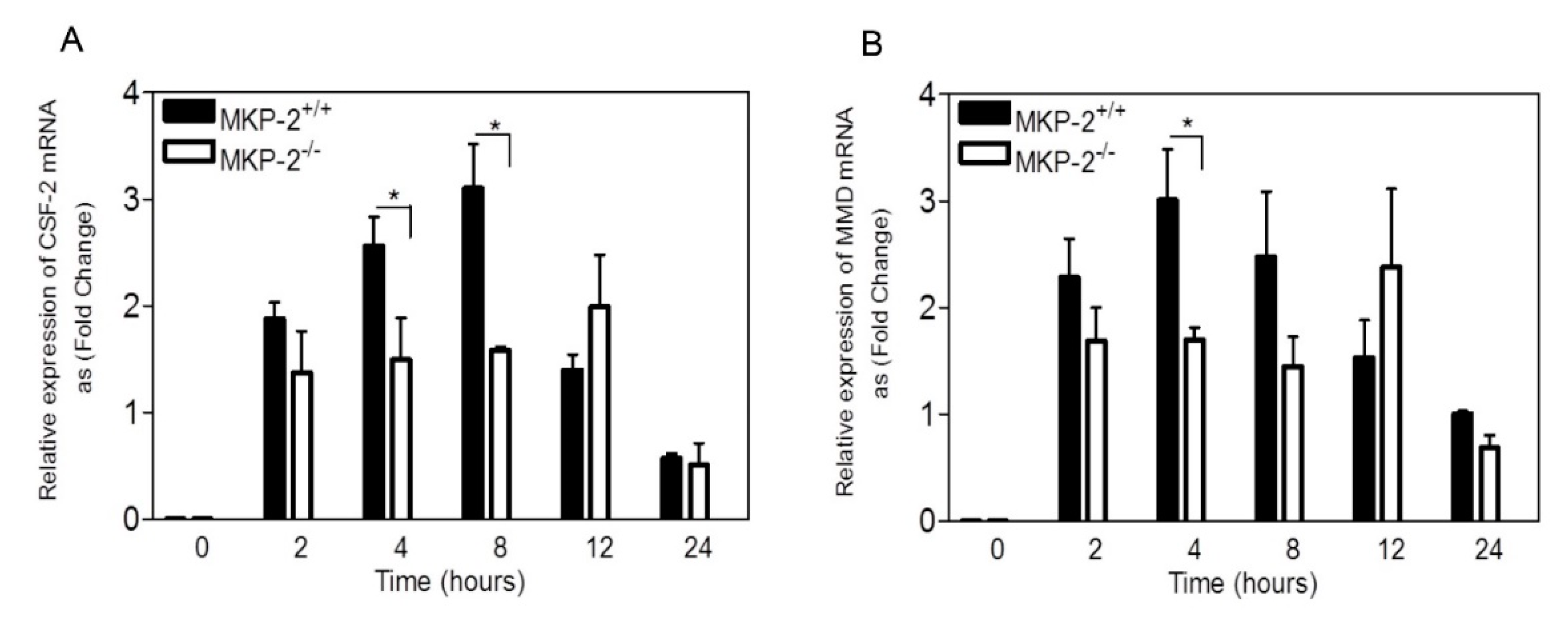
2. Results
2.1. The Effect of MKP-2 in the Gene Expression Pattern of LPS-Stimulated Macrophages
2.2. The Role of MKP-2 in the Expression of M-CSF-Induced Macrophage Differentiation and Proliferation Genes
2.3. MKP-2 Deletion Negatively Effects M-CSF Stimulated Macrophage Proliferation
2.4. MKP-2 Deletion Enhances ERK Kinase Signaling
2.5. The Effect of ERK Inhibition on M-CSF Induced Proliferation and Mmd and Csf2 Expression
2.6. Differential Expression of CD34 and CD115 on the Surface of Bone Marrow Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. DUSP-4 Deletion Mice
4.3. Cell Culture
4.4. Microarray Expression Analysis
4.5. Quantitative Real-Time Polymerase Chain Reaction Analysis
4.6. Flow Cytometry Assay
4.7. Proliferation Assay
4.7.1. Cell Counting by Hematoxylin
4.7.2. BrdU Cell Proliferation Assay
4.8. Western Blotting
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rincon, M.; Pedraza-Alva, G. JNK and p38 MAP kinases in CD4+ and CD8+ T cells. Immunol. Rev. 2003, 192, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.; Mages, J.; Dietrich, H.; Servatius, A.; Howells, N.; Cato, A.C.; Lang, R. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 2006, 203, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Salojin, K.V.; Owusu, I.B.; Millerchip, K.A.; Potter, M.; Platt, K.A.; Oravecz, T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 2006, 176, 1899–1907. [Google Scholar] [CrossRef]
- Jeffrey, K.L.; Brummer, T.; Rolph, M.S.; Liu, S.M.; Callejas, N.A.; Grumont, R.J.; Gillieron, C.; Mackay, F.; Grey, S.; Camps, M.; et al. Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat. Immunol. 2006, 7, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Blattman, J.N.; Kennedy, N.J.; Duong, J.; Nguyen, T.; Wang, Y.; Davis, R.J.; Greenberg, P.D.; Flavell, R.A.; Dong, C. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 2004, 430, 793–797. [Google Scholar] [CrossRef]
- Sloss, C.M.; Cadalbert, L.; Finn, S.G.; Fuller, S.J.; Plevin, R. Disruption of two putative nuclear localization sequences is required for cytosolic localization of mitogen-activated protein kinase phosphatase-2. Cell Signal 2005, 17, 709–716. [Google Scholar] [CrossRef]
- Chen, P.; Hutter, D.; Yang, X.; Gorospe, M.; Davis, R.J.; Liu, Y. Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J. Biol. Chem. 2001, 276, 29440–29449. [Google Scholar] [CrossRef]
- Lawan, A.; Torrance, E.; Al-Harthi, S.; Al-Harthi, M.; Alnasser, S.; Neamatallah, T.; Schroeder, J.; Plevin, R. MKP-2: Out of the DUSP-bin and back into the limelight. Biochem. Soc. Trans. 2012, 40, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, M.S.; Cadalbert, L.C.; McGachy, H.A.; Shweash, M.; Schroeder, J.; Kurnik, M.; Sloss, C.M.; Bryant, C.E.; Alexander, J.; Plevin, R. MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana. PLoS Pathog. 2010, 6, e1001192. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.; McGachy, H.A.; Woods, S.; Plevin, R.; Alexander, J. T cell hypo-responsiveness against Leishmania major in MAP kinase phosphatase (MKP) 2 deficient C57BL/6 mice does not alter the healer disease phenotype. PLoS Negl. Trop. Dis. 2013, 7, e2064. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.; Schroeder, J.; McGachy, H.A.; Plevin, R.; Roberts, C.W.; Alexander, J. MAP kinase phosphatase-2 plays a key role in the control of infection with Toxoplasma gondii by modulating iNOS and arginase-1 activities in mice. PLoS Pathog. 2013, 9, e1003535. [Google Scholar] [CrossRef]
- Cornell, T.T.; Rodenhouse, P.; Cai, Q.; Sun, L.; Shanley, T.P. Mitogen-activated protein kinase phosphatase 2 regulates the inflammatory response in sepsis. Infect. Immun. 2010, 78, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Lin, Y.C.; Hsiao, W.Y.; Liao, F.H.; Huang, P.Y.; Tan, T.H. DUSP4 deficiency enhances CD25 expression and CD4+ T-cell proliferation without impeding T-cell development. Eur. J. Immunol. 2012, 42, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Sharif-Askari, E.; Vassen, L.; Kosan, C.; Khandanpour, C.; Gaudreau, M.C.; Heyd, F. Zinc finger protein Gfi1 controls the endotoxin-mediated Toll-like receptor inflammatory response by antagonizing NF-kappaB p65. Mol. Cell. Biol. 2010, 30, 3929–3942. [Google Scholar] [CrossRef] [PubMed]
- Morstyn, G.; Burgess, A.W. Hemopoietic growth factors: A review. Cancer Res. 1988, 48, 5624–5637. [Google Scholar] [PubMed]
- Francke, A.; Herold, J.; Weinert, S.; Strasser, R.H.; Braun-Dullaeus, R.C. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. J. Histochem. Cytochem. 2011, 59, 813–825. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ebihara, Y.; Xu, M.J.; Ishii, T.; Sugiyama, D.; Yoshino, H.; Nakahata, T. CD34 expression on long-term repopulating hematopoietic stem cells changes during developmental stages. Blood 2001, 97, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Björkbacka, H.; Fitzgerald, K.A.; Huet, F.; Li, X.; Gregory, J.A.; Lee, M.A.; Freeman, M.W. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol. Genom. 2004, 19, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Shuyi, Z.; Kim, C.C.; Batra, S.; McKerrow, J.H.; Loke, P.N. Delineation of Diverse Macrophage Activation Programs in Response to Intracellular Parasites and Cytokines. PLoS Negl. Trop. Dis. 2010, 4, e648. [Google Scholar]
- Sikkeland, L.I.B.; Dahl, C.P.; Ueland, T.; Andreassen, A.K.; Gude, E.; Edvardsen, T.; Aukrust, P. Increased levels of inflammatory cytokines and endothelin-1 in alveolar macrophages from patients with chronic heart failure. PLoS ONE 2012, 7, e36815. [Google Scholar] [CrossRef] [PubMed]
- Wahl, J.R.; Goetsch, N.J.; Young, H.J.; Maanen, R.J.V.; Johnson, J.D.; Pea, A.S.; Brittingham, A. Murine macrophages produce endothelin-1 after microbial stimulation. Exp. Biol. Med. (Maywood) 2005, 230, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Javeshghani, D.; Barhoumi, T.; Idris-Khodja, N.; Paradis, P.; Schiffrin, E.L. Reduced Macrophage-Dependent Inflammation Improves Endothelin-1–Induced Vascular Injury. Hypertension 2013, 62, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Elisa, T.; Antonio, P.; Giuseppe, P.; Alessandro, B.; Giuseppe, A.; Federico, C.; Daniela, R. Endothelin Receptors Expressed by Immune Cells Are Involved in Modulation of Inflammation and in Fibrosis: Relevance to the Pathogenesis of Systemic Sclerosis. J. Immunol. Res. 2015, 2015, 147616. [Google Scholar] [CrossRef]
- Freeman, B.D.; Machado, F.S.; Tanowitz, H.B.; Desruisseaux, M.S. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. 2014, 118, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehli, M.; Krause, S.W.; Schwarzfischer, L.; Kreutz, M.; Andreesen, R. Molecular cloning of a novel macrophage maturation-associated transcript encoding a protein with several potential transmembrane domains. Biochem. Biophys. Res. Commun. 1995, 217, 661–667. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, J.; Yin, D.D.; Xiang, J.; He, F.; Wang, Y.C.; Han, H. Monocyte to macrophage differentiation-associated (MMD) positively regulates ERK and Akt activation and TNF-α and NO production in macrophages. Mol. Biol. Rep. 2012, 39, 5643–5650. [Google Scholar] [CrossRef]
- Robertson, S.A.; Sjoblom, C.; Jasper, M.J.; Norman, R.J.; Seamark, R.F. Granulocyte-Macrophage Colony-Stimulating Factor Promotes Glucose Transport and Blastomere Viability in Murine Preimplantation Embryos. Biol. Reprod. 2001, 64, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Al-Harthi, S.; Cadalbert, L.; McCluskey, A.G.; Shweash, M.; Grassia, G.; Plevin, R. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J. Biol. Chem. 2011, 286, 12933–12943. [Google Scholar] [CrossRef] [PubMed]
- Jaworowski, A.; Christy, E.; Yusoff, P.; Byrne, R.; Hamilton, J.A. Differences in the kinetics of activation of protein kinases and extracellular signal-related protein kinase 1 in colony-stimulating factor 1-stimulated and lipopolysaccharide-stimulated macrophages. Biochem. J. 1996, 320, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworowski, A.; Wilson, N.J.; Christy, E.; Byrne, R.; Hamilton, J.A. Roles of the mitogen-activated protein kinase family in macrophage responses to colony stimulating factor-1 addition and withdrawal. J. Biol. Chem. 1999, 274, 15127–15133. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. CSF-1 signal transduction. J. Leukoc. Biol. 1997, 62, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.T.; Craggs, G.; Wilson, L.; Kellie, S. Mechanism of phosphatidylinositol 3-kinase-dependent increases in BAC1.2F5 macrophage-like cell density in response to M-CSF: Phosphatidylinositol 3-kinase inhibitors increase the rate of apoptosis rather than inhibit DNA synthesis. Inflamm. Res. 2000, 49, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Francis, M.K.; Lorenzini, A.; Tresini, M.; Cristofalo, V.J. Metabolic stabilization of MAP kinase phosphatase-2 in senescence of human fibroblasts. Exp. Cell Res. 2003, 290, 195–206. [Google Scholar] [CrossRef]
- Valledor, A.F.; Comalada, M.; Xaus, J.; Celada, A. The Differential Time-course of Extracellular-regulated Kinase Activity Correlates with the Macrophage Response toward Proliferation or Activation. J. Biol. Chem. 2000, 275, 7403–7409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, E.T.; Shukla, S.; Nagy, N.; Boom, W.H.; Beck, R.C.; Zhou, L.; Harding, C.V. ERK Signaling Is Essential for Macrophage Development. PLoS ONE 2015, 10, e0140064. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Yamamoto, T.; Tsuchiya, Y.; Nishida, E. ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci. 2006, 97, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Comalada, M.; Farrera, C.; Valledor, A.F.; Lloberas, J.; Celada, A. Macrophage-colony-stimulating factor-induced proliferation and lipopolysaccharide-dependent activation of macrophages requires Raf-1 phosphorylation to induce mitogen kinase phosphatase-1 expression. J. Immunol. 2006, 176, 6594–6602. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Xaus, J.; Marquès, L.; Celada, A. Macrophage colony-stimulating factor induces the expression of mitogen-activated protein kinase phosphatase-1 through a protein kinase C-dependent pathway. J. Immunol. 1999, 163, 2452–2462. [Google Scholar] [PubMed]
- Valledor, A.F.; Arpa, L.; Sánchez-Tilló, E.; Comalada, M.; Casals, C.; Xaus, J.; Celada, A. IFN-{gamma}-mediated inhibition of MAPK phosphatase expression results in prolonged MAPK activity in response to M-CSF and inhibition of proliferation. Blood 2008, 112, 3274–3282. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-W.; Kang, T.H.; Kim, W.; Choi, Y.H.; Kim, K.T. Mitogen-activated protein kinase phosphatase 2 regulates histone H3 phosphorylation via interaction with vaccinia-related kinase 1. Mol. Biol. Cell 2013, 24, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Accart, N.; Kintz, J.; Cochin, S.; Reymann, C.; Le Pogam, F.; Fournel, S. Therapeutic effects of anti-CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor-associated macrophages and osteoclasts. PLoS ONE 2013, 8, e73310. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Colony-stimulating factor-1 receptor. Blood 1990, 75, 1–12. [Google Scholar] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. 2), S3–S23. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.K.; Narni-Mancinelli, E.; Senechal, B.; Trouillet, C.; Saederup, N.; Molina, T. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med. 2009, 206, 595–606. [Google Scholar] [CrossRef]
- Jacome-Galarza, C.E.; Lee, S.K.; Lorenzo, J.A.; Aguila, H.L. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J. Bone Miner. Res. 2013, 28, 1203–1213. [Google Scholar] [CrossRef]
- Barbour, M.; Plevin, R.; Jiang, H.-R. MAP kinase phosphatase 2 deficient mice develop attenuated experimental autoimmune encephalomyelitis through regulating dendritic cells and T cells. Sci. Rep. 2016, 6, 38999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Iersel, M.P.; Kelder, T.; Pico, A.R.; Hanspers, K.; Coort, S.; Conklin, B.R.; Evelo, C. Presenting and exploring biological pathways with PathVisio. BMC Bioinf. 2008, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Sturn, A. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | FC-MKP-2+/+ | FC-MKP-2-/- | Gene Symbol | Gene Name |
|---|---|---|---|---|
| 1 | 4717.09 | 3292.84 | Il19 | interleukin 19 |
| 2 | 2975.03 | 1604.87 | Cxcl1 | chemokine (C-X-C motif) ligand 1 |
| 3 | 1850.59 | 1746.34 | Ptgs2 | prostaglandin-endoperoxide synthase 2 |
| 4 | 1758.98 | 1369.49 | Il1α | interleukin 1 alpha |
| 5 | 1574.15 | 663.96 | Gfi1 | growth factor independent 1 |
| 6 | 1094.3 | 5176.78 | Edn1 | endothelin 1 |
| 7 | 769.02 | 742.88 | Il6 | interleukin 6 |
| 8 | 710.97 | 446.13 | Cxcl2 | chemokine (C-X-C motif) ligand 2 |
| 9 | 434.82 | 425.43 | Dusp2 | dual specificity phosphatase 2 |
| 10 | 423.49 | 494.97 | Il1β | interleukin 1 beta |
| GO Term | Description | p-Value |
|---|---|---|
| GO:0009611 | response to wounding | 4.10E−14 |
| GO:0050727 | regulation of inflammatory response | 1.97E−13 |
| GO:0001817 | regulation of cytokine production | 2.75E−13 |
| GO:0051239 | regulation of multicellular organismal process | 2.07E−12 |
| GO:0032101 | regulation of response to external stimulus | 1.42E−11 |
| GO:0048518 | positive regulation of biological process | 4.44E−11 |
| GO:0023051 | regulation of signalling | 6.00E−11 |
| GO:0010646 | regulation of cell communication | 8.51E−11 |
| GO:0008284 | positive regulation of cell proliferation | 9.07E−11 |
| GO:0050865 | regulation of cell activation | 1.63E−10 |
| GO:0010941 | regulation of cell death | 1.32E−09 |
| GO:0008009 | chemokine activity | 8.31E−08 |
| GO:0032496 | response to lipopolysaccharide | 1.09E−08 |
| GO:0001818 | negative regulation of cytokine production | 1.50E−08 |
| GO:0045595 | regulation of cell differentiation | 7.88E−08 |
| GO:0032680 | regulation of tumor necrosis factor production | 1.06E−07 |
| GO:0042625 | ATPase activity, coupled to transmembrane movement of ions | 5.32E−05 |
| GO:0017017 | MAP kinase tyrosine/serine/threonine phosphatase activity | 3.86E−04 |
| Primer Name | Sequence | PCR Product Size (bp) |
|---|---|---|
| Qars | Forward: 5′-GGACTCCAGCTGAGCGCTGCTC-3′ Reverse: 5′-GGTGGACTCCACAGCTTCCTCAAT-3′ | 138 |
| Csf-2 | Forward: 5′-TGCCTGTCACGTTGAATGAAGAGG-3′ Reverse: 5′-TGTCTGGTAGTAGCTGGCTGTCATGTTC-3′ | 164 |
| Mmd | Forward: 5′-TGGCCGCTACAAACCAACGTG-3′ Reverse: 5′-CAAAGGCCCATCCCGTAGATCC-3′ | 156 |
| Edn1 | Forward: 5′- ACA CTC CCG AGC GCG TCG TA -3′ Reverse: 5′- TCT TGT CTT TTT GGT GAG CGC ACT G -3′ | 142 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neamatallah, T.; Jabbar, S.; Tate, R.; Schroeder, J.; Shweash, M.; Alexander, J.; Plevin, R. Whole Genome Microarray Analysis of DUSP4-Deletion Reveals A Novel Role for MAP Kinase Phosphatase-2 (MKP-2) in Macrophage Gene Expression and Function. Int. J. Mol. Sci. 2019, 20, 3434. https://doi.org/10.3390/ijms20143434
Neamatallah T, Jabbar S, Tate R, Schroeder J, Shweash M, Alexander J, Plevin R. Whole Genome Microarray Analysis of DUSP4-Deletion Reveals A Novel Role for MAP Kinase Phosphatase-2 (MKP-2) in Macrophage Gene Expression and Function. International Journal of Molecular Sciences. 2019; 20(14):3434. https://doi.org/10.3390/ijms20143434
Chicago/Turabian StyleNeamatallah, Thikryat, Shilan Jabbar, Rothwelle Tate, Juliane Schroeder, Muhannad Shweash, James Alexander, and Robin Plevin. 2019. "Whole Genome Microarray Analysis of DUSP4-Deletion Reveals A Novel Role for MAP Kinase Phosphatase-2 (MKP-2) in Macrophage Gene Expression and Function" International Journal of Molecular Sciences 20, no. 14: 3434. https://doi.org/10.3390/ijms20143434