Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease


Laboratory of Lipid Biochemistry and Protein Interactions, Department of Cellular and Molecular Medicine, KU Leuven—University of Leuven, 3000 Leuven, Belgium
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(15), 3673; https://doi.org/10.3390/ijms20153673
Submission received: 28 June 2019
/
Revised: 24 July 2019
/
Accepted: 25 July 2019
/
Published: 26 July 2019
(This article belongs to the Special Issue Peroxisomes under the Spotlight: Collaboration is the Way to Go)
Abstract
:Hydrogen peroxide (H2O2), a non-radical reactive oxygen species generated during many (patho)physiological conditions, is currently universally recognized as an important mediator of redox-regulated processes. Depending on its spatiotemporal accumulation profile, this molecule may act as a signaling messenger or cause oxidative damage. The focus of this review is to comprehensively evaluate the evidence that peroxisomes, organelles best known for their role in cellular lipid metabolism, also serve as hubs in the H2O2 signaling network. We first briefly introduce the basic concepts of how H2O2 can drive cellular signaling events. Next, we outline the peroxisomal enzyme systems involved in H2O2 metabolism in mammals and reflect on how this oxidant can permeate across the organellar membrane. In addition, we provide an up-to-date overview of molecular targets and biological processes that can be affected by changes in peroxisomal H2O2 metabolism. Where possible, emphasis is placed on the molecular mechanisms and factors involved. From the data presented, it is clear that there are still numerous gaps in our knowledge. Therefore, gaining more insight into how peroxisomes are integrated in the cellular H2O2 signaling network is of key importance to unravel the precise role of peroxisomal H2O2 production and scavenging in normal and pathological conditions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Hydrogen peroxide (H2O2), the non-radical 2-electron reduction product of oxygen, is a natural metabolite commonly found in aerobic organisms [1]. For a long time, this compound was considered as an unwanted and rather detrimental by-product of oxidative metabolism [2]. However, during the last decades, H2O2 has moved into the forefront as a central redox signaling molecule in many biological processes such as cell proliferation and differentiation, tissue repair, inflammation, circadian rhythm, and aging [1,3]. The signaling properties of H2O2 can be attributed to its relative stability, diffusibility, and selective reactivity (see Section 2). However, whether H2O2 acts as a signaling molecule or leads to oxidative damage of biomolecules, a condition denoted as oxidative stress, depends on the cellular context, its local concentration, and the kinetics of its production and elimination [4]. For example, in the presence of free redox-active metal ions, non-toxic physiological levels of H2O2 can give rise to highly toxic hydroxyl radicals (●OH), which indiscriminately oxidize virtually any organic molecule they encounter [5,6].
Under steady state conditions, the in cellulo production and consumption of H2O2 are balanced. Major metabolic sources of H2O2 include the flavin-dependent oxidases (e.g., the endoplasmic reticulum oxidoreductase 1 (ERO1) [7]; the acyl-coenzyme A (acyl-CoA) oxidases in peroxisomes (see Section 3.1)); and superoxide dismutases (SODs) [8]. The latter group of enzymes catalyzes the dismutation of superoxide anion radicals (O2●−), which are predominantly produced by the mitochondrial electron transport chain [9] and membrane-associated NADPH oxidases (NOXs) that are located in various subcellular compartments [10]. Major H2O2-scavenging enzymes include catalase (CAT) and various thiol-based peroxidases such as glutathione peroxidases (GPXs) and peroxiredoxins (PRDXs) [11]. While CAT is predominantly located in peroxisomes (see Section 3.2), GPXs [12] and PRDXs [13] are often localized in various subcellular compartments.
Peroxisomes are cell organelles that are best-known for their involvement in cellular lipid metabolism [14]. In mammals, this entails the α- and β-oxidation of fatty acids and the biosynthesis of ether-phospholipids, bile acids, and docosahexaenoic acid [15]. Other metabolic functions of peroxisomes in mammals include glyoxylate detoxification, amino acid catabolism, polyamine oxidation, and the production and scavenging of reactive oxygen and nitrogen species (ROS and RNS, respectively) [16,17]. In addition, these organelles are also increasingly recognized as important hubs in innate immune-, lipid-, inflammatory-, and redox-signaling networks [18]. To perform these functions, peroxisomes can dynamically regulate their number, shape, and protein content in response to changing environmental conditions [19]. They also have to stay in close communication with other subcellular compartments [20]. The formation and maintenance of peroxisomes requires a specialized set of proteins, called peroxins (acronym: PEX; the number refers to their order of discovery) [21], and superfluous and dysfunctional organelles are targeted for lysosomal degradation through a process known as pexophagy [22]. Alterations in any of these metabolic, signaling, or other pathways have been linked to multiple genetic (e.g., Zellweger syndrome) [23], infectious [24], and oxidative stress-related (e.g., neurodegeneration, diabetes, and cancer) diseases [25,26]. In the following sections, we focus on the role of mammalian peroxisomes in cellular H2O2 metabolism and how perturbations in this process may affect cellular function and organismal health.
2. The Basic Concepts of H2O2 Signaling
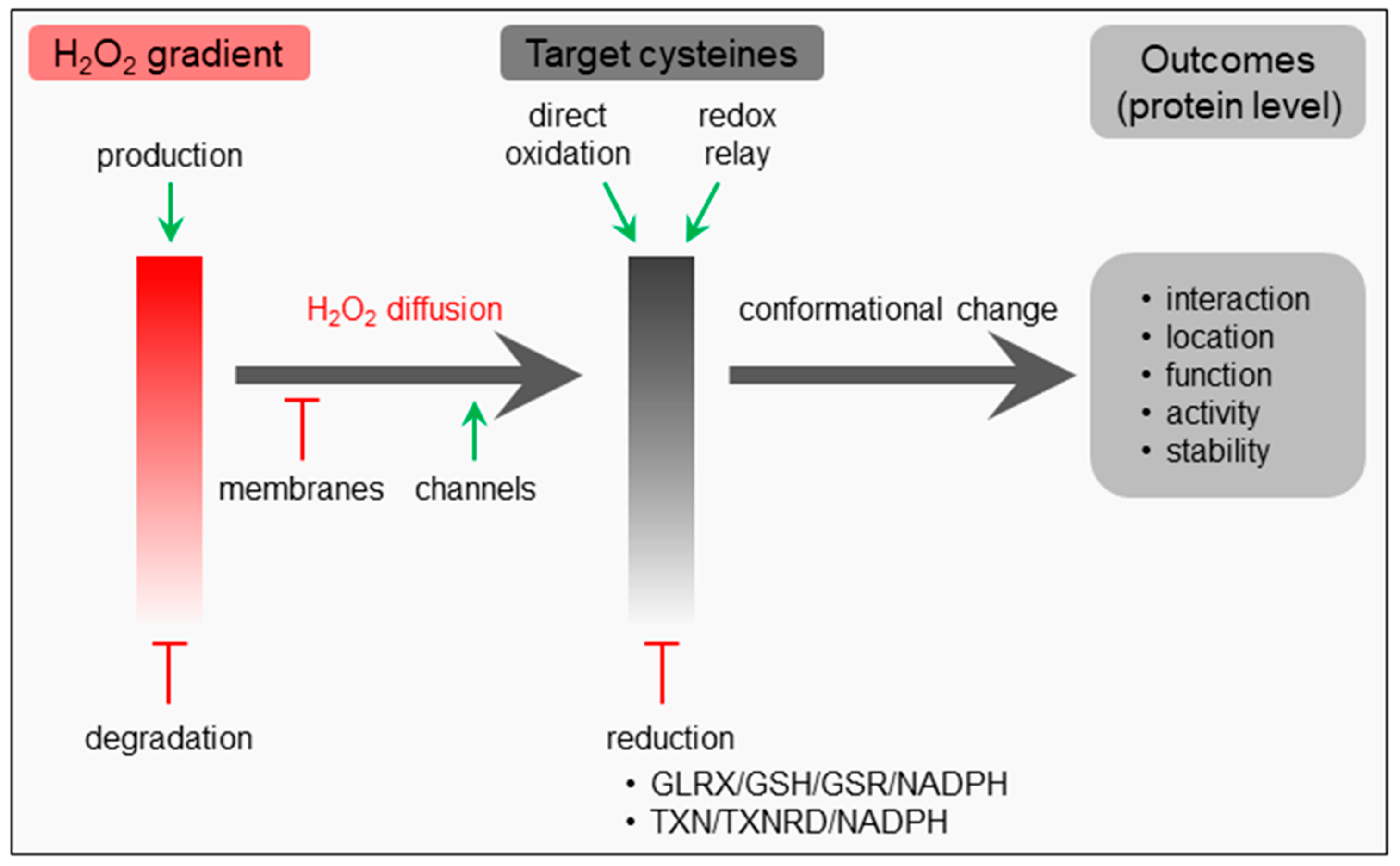
Like many other signaling pathways, hallmarks of H2O2 signaling include messenger formation, messenger metabolism, messenger action, and recovery [27]. Given its relative stability (cellular half-life: ~1 ms), diffusibility, and selective reactivity, H2O2 is often put forth as the most important redox signaling molecule [1,28,29]. This non-radical ROS can be formed and degraded in different subcellular compartments (see Section 1), and its local concentration depends on its rates of synthesis, degradation, and diffusion (Figure 1). Importantly, although H2O2 is a non-charged molecule, its dipole moment is higher than that of water, thereby severely limiting its transport across lipid membranes by simple diffusion [29]. Indeed, efficient permeation of this molecule across biological membranes requires specific channel proteins, referred to as peroxiporins [1]. The spatial separation of sources and sinks as well as its diffusion away from the site of production and across biomembranes lead to the formation of intracellular H2O2 gradients, which determine the behavior of responsive systems [6].
The primary messenger action of H2O2 depends on its ability to oxidize a variety of target proteins with a high degree of specificity, predominantly through reaction with nucleophilic cysteine thiolate groups (Cys-S−) that can be found in specific protein microenvironments [1]. Oxidation of such deprotonated cysteine residues can lead to the formation of unstable sulfenic acid (-SOH) intermediates that can (i) be reduced again, (ii) react with other proximal thiol groups to form intra- or intermolecular disulfide bonds, or (iii) undergo hyperoxidation to form sulfinic (-SO2H) and then sulfonic (-SO3H) acids [11,30]. Disulfide bond formation can induce conformational changes, leading to alterations in macromolecular interactions, protein localization, function, activity, and/or stability; and protein disulfides can be converted back to their reduced state by components of the glutaredoxin (GLRX)/glutathione (GSH)/glutathione-disulfide reductase (GSR)- and thioredoxin (TXN)/thioredoxin reductase (TXNRD)-containing antioxidant systems (Figure 1) [31,32]. As such, oxidation-susceptible cysteine thiols can function as regulatory switches that transmit information along a signaling cascade after being oxidized by H2O2 [11].
Importantly, depending on the context and circumstances (e.g., the subcellular location, the H2O2 concentration, and duration of exposure, etc.), the oxidation of signaling proteins may occur through direct reaction of H2O2 with hyper-reactive thiols or indirectly through thioredoxin- or peroxiredoxin- catalyzed redox relay reactions (Figure 1) [33,34]. This implies that the thiol peroxidases involved may display a dual function: on one hand, they can function as H2O2 scavengers that counteract redox signaling; on the other hand, they may act as enablers of protein thiol oxidation by transferring oxidative equivalents from H2O2 to redox-regulated proteins [34]. In the latter mechanism, the target specificity is likely to be determined by protein–protein interactions. In this context, it is relevant to mention that the reducing power required to maintain thiol redox networks is provided by NADPH [35].
To illustrate the complexity of H2O2-mediated signaling; we briefly focus on one specific example: the activity modulation of nuclear factor erythroid 2-related factor 2 (NFE2L2). NFE2L2, also known as NRF2, is a transcription factor regulating the expression of genes containing an antioxidant/electrophile response element motif in their promoter [36]. Under homeostatic conditions, NFE2L2 is constitutively degraded by different pathways: on one hand, the protein interacts with Kelch-like ECH-associated protein 1 (KEAP1), a redox-regulated substrate adapter protein of the cullin 3-dependent E3 ubiquitin ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of KEAP1-associated proteins; on the other hand, the protein can be phosphorylated by glycogen synthase kinase 3β (GSK-3β), thereby creating a phosphodegron that drives protein degradation through interaction with a ubiquitin ligase [36,37]. However, upon exposure to H2O2, specific cysteine residues of KEAP1 (e.g., Cys151, Cys171, Cys273, and Cys288 in the human protein) are oxidized, thereby inducing conformational changes interfering with the ability of KEAP1 to ubiquitinate NFE2L2 [38]. In addition, given that (i) phosphorylation of GSK-3β at serine 9 inhibits the kinase’s activity [39], and (ii) multiple kinases (e.g., the serine/threonine protein kinases B and C, the mitogen-activated protein kinases 1 and 14) that can mediate this phosphorylation event are activated by H2O2 [36], such condition also results in a KEAP1-independent activation of NFE2L2. Importantly, electrophiles such as H2O2 can also modulate the activity of NFE2L2 at other levels. For example, they can (i) enhance the translation rate of the NFE2L2 mRNA [40,41], (ii) stimulate the nuclear accumulation of the NFE2L2 by freeing a nuclear localization sequence that is not surface-exposed in the KEAP1-bound transcription factor and/or by oxidatively modifying (or masking) nuclear export sequences in KEAP1 or the NFE2L2–KEAP1 complex [36], (iii) increase the acetylation state of NFE2L2, a condition reported to correlate with DNA binding and transcription factor activity [42,43], and (iv) affect the interaction of NFE2L2 with its transcriptional regulators [36].
3. Players in Peroxisomal H2O2 Metabolism and Transport
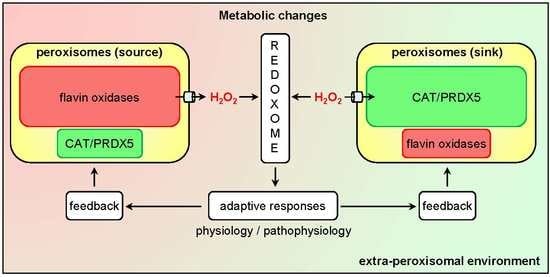
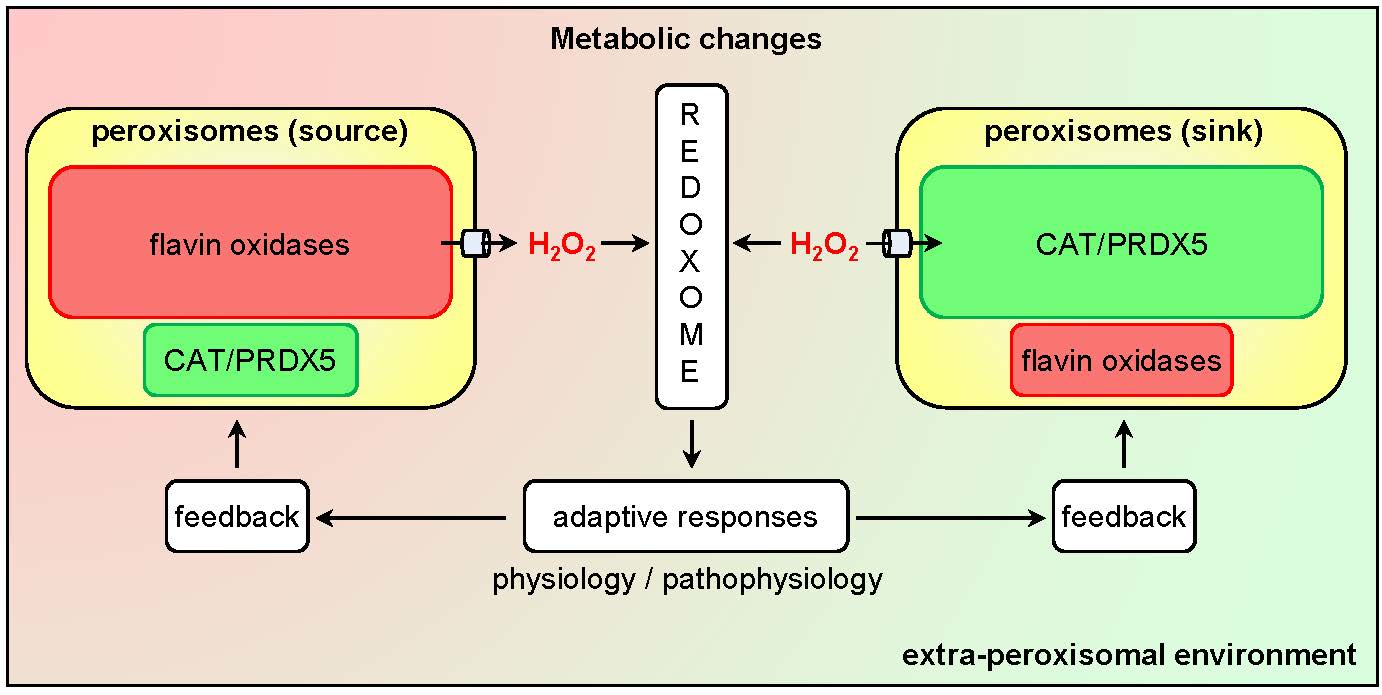
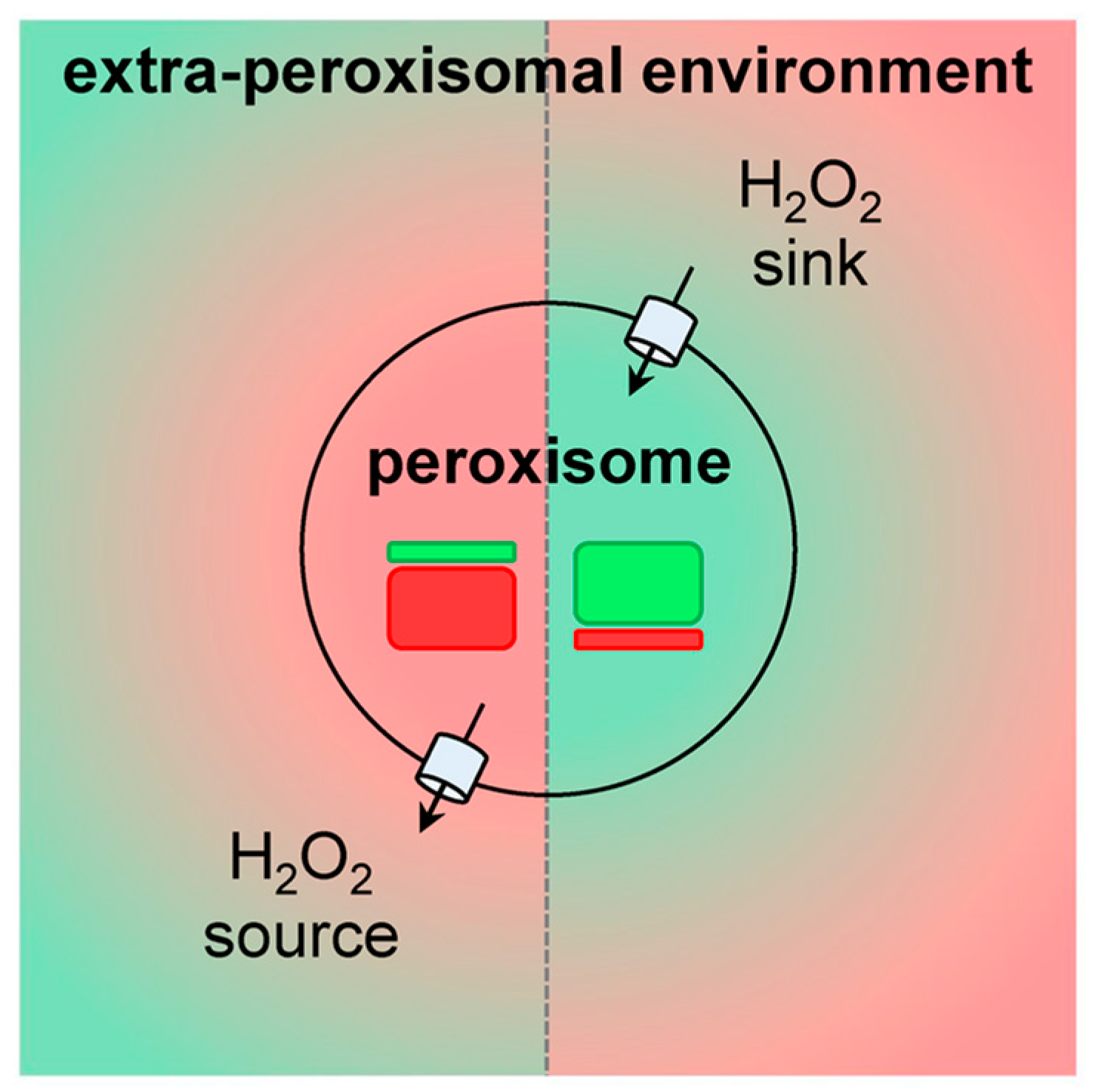
Currently, it is clear that peroxisomes contain both enzymes that can produce (see Section 3.1) or scavenge (see Section 3.2) H2O2. In addition, there is compelling evidence that the peroxisomal membrane contains non-selective pore-forming proteins that allow the exchange of small metabolites (see Section 3.3). As such, these organelles can be expected to influence local H2O2 gradients and, therefore, H2O2-mediated signaling events. Whether peroxisomes act as a net sinks or sources of H2O2 (Figure 2) most likely depends on the cell type, its physiological state, and the microenvironment.
3.1. H2O2-Generating Systems
Peroxisomes are metabolically active organelles involved in a variety of biochemical processes (see Section 1). A specific subset of enzymes acting in these processes include the flavin adenine dinucleotide (FAD)- or flavin mononucleotide (FMN)-dependent oxidases (for a detailed list, see [44]), of which the reduced flavin forms are regenerated by reduction of molecular oxygen (O2) to H2O2. In this context, it is important to note that (i) unlike mitochondria, peroxisomes lack a respiratory chain [45], and (ii) depending on the oxidase, the cofactor can be loosely or firmly bound [46,47]. Natural substrates of the mammalian H2O2-producing peroxisomal oxidases include, among others, different types of fatty acids (e.g., very-long-chain fatty acids, 2-methyl-branched fatty acids, 2-hydroxy fatty acids, and bile acid intermediates) [48], d-amino acids (e.g., d-Ser) [49], polyamines (e.g., N1-acetylspermine) [50], glycolate [51], and pipecolic acid [52]. Depending on the activity of these oxidases, which may be regulated by numerous posttranslational modifications (e.g., phosphorylation, acetylation, ubiquitination, succinylation, mono- or di-methylation [53]), and the intraperoxisomal H2O2 scavenging rates (see Section 3.2), changes in substrate availability or enzyme activity may result in altered H2O2 levels. This is nicely illustrated by a recent study showing that (i) lysine succinylation stimulates the activity of acyl-CoA oxidase 1 (ACOX1), a rate-limiting enzyme in peroxisomal fatty acid β-oxidation, (ii) sirtuin 5 (SIRT5), an NAD-dependent protein lysine desuccinylase, can locate to peroxisomes where it binds to and desuccinylates ACOX1, and (iii) downregulation of SIRT5 increases ACOX1 activity, peroxisomal H2O2 production, and oxidative DNA damage in cultured HepG2 liver cells, mouse livers, and/or human hepatocellular carcinoma samples [54].
3.2. H2O2-Elimination Systems
Currently, it is clear that mammalian peroxisomes are equipped with at least two H2O2-eliminating enzyme systems, of which the best characterized enzyme is CAT. Depending on the H2O2 concentration and the presence of other metabolic hydrogen donors (AH2) such as short-chain aliphatic alcohols, formate, or nitrite, this heme-containing protein can scavenge H2O2 in a catalatic (2 H2O2 → 2 H2O + O2) or peroxidatic (H2O2 + AH2 → A + 2 H2O) manner [55]. In contrast to other H2O2-decomposing enzymes, these reactions occur without the use of reducing equivalents [56]. CAT is an abundant, predominantly peroxisomal enzyme. However, depending on the cell type and environmental conditions, the protein may also be (partially) localized to the cytosol and nucleus [57,58]. In this context, it is worth noting that oxidative stress impairs the import efficiency of CAT into peroxisomes, a phenomenon that apparently protects the cytosol against H2O2-induced insults [59]. In addition, upon exposure of mammalian cells to external H2O2 or other ROS stimuli, (cytosolic) catalase can be phosphorylated by protein kinase C delta (PRKCD) at Ser167 [60] and by the Abelson tyrosin-protein kinases ABL1 and ABL2 at both Tyr231 and Tyr386 [61], thereby enhancing its activity. On the other hand, the activity of CAT can be down-regulated through nitrosylation of Cys377 [62] and S-thiolation at not yet identified cysteine residues [63,64]. Finally, CAT can also be phosphorylated [65], acetylated [66], succinylated [54,67], monomethylated [68,69], ubiquitinated [70,71], and sumoylated [72] on many other residues. However, how these posttranslational modification events affect CAT localization and/or activity remains to be determined. From these observations, combined with the fact that CAT is not essential for life [73,74], it is apparent that more studies are needed to fully understand the precise regulation and physiological function of this H2O2-scavenging enzyme in peroxisomal redox biology.
Another sink for H2O2 inside peroxisomes is PRDX5, a thiol-dependent monomeric peroxidase that is also located in the cytosol, the nucleus, and mitochondria [75]. PRDX5 can also reduce peroxynitrite (ONOO−) and a variety of lipid peroxides (LOOH) [76]. Currently, it is widely accepted that PRDX5 uses an NADPH-dependent thioredoxin (TXN)/TXN reductase (TXNTR) system to reduce its substrates [31]. Intriguingly, no such enzyme system has yet been identified in mammalian peroxisomes. Like CAT, PRDX5 can also be phosphorylated [77,78], acetylated [66,79], succinylated [67], glutathionylated [80], and ubiquitinated [70,71]. In addition, the protein can undergo disulfide bond formation [81,82]. Once again, how these posttranslational modifications affect PRDX5 activity remains to be clarified. The physiological function of peroxisomally located PRDX5 is also not yet clear. On one hand, the protein may act as an antioxidant by inactivating H2O2 before it can modify redox-sensitive cysteines [83]. On the other hand, it may act as a redox relay factor by transferring oxidizing equivalents from H2O2 to target proteins through thiol-disulfide reshuffling [33,34]. In the latter case, the peroxisomal pools of PRDX5 and CAT may play non-overlapping roles in H2O2 clearance, a paradigm supported by the observation that both antioxidant enzymes present distinct kinetic characteristics. Indeed, PRDX5 and CAT scavenge H2O2 in the low micromolar and low millimolar range, respectively, and the maximum rate of H2O2 removal is orders of magnitude more for CAT than for PRDX5 [76,84].
Besides CAT and PRDX5, H2O2 may also be removed by glutathione peroxidases (GPXs), a family of antioxidant enzymes that typically use GSH as reductant [85]. However, despite the fact that rat liver peroxisomes appear to contain GPX activity [86], no such enzyme has yet been identified at the protein level.
3.3. H2O2 Permeation across the Peroxisomal Membrane
Throughout the years, it has become clear the biological membranes, including the peroxisomal one, act as permeability barriers for H2O2 [87,88]. Importantly, to serve as a subcellular platform for H2O2 signaling, it is essential that peroxisomes can exchange this redox messenger with their environment. That this is indeed the case, has already been demonstrated in vitro [89,90], in cellulo [91], and in slices of liver, lung, and lenses from CAT-deficient mice [92]. In general, it is thought that this permeation process is governed by peroxiporins (see Section 2), a class of proteins that has not yet been assigned to peroxisomes. However, here it is important to note that the peroxisomal membrane contains a non-selective pore-forming protein, termed PXMP2, that allows free diffusion of small molecules (< 300–600 Da) [93]. Nonetheless, we have recently demonstrated that neither this peroxisomal membrane protein (PMP) nor PEX11B, another widely expressed PMP whose yeast homologue enables the permeation of molecules up to 400 Da [94,95], are essential for the transport of H2O2 across the peroxisomal membrane [96].
4. The Emerging Roles of Peroxisomes in Cellular H2O2 Signaling
A general requisite for signal propagation is the spatial segregation of opposing reactions, a condition resulting in the formation of concentration gradients [97]. Therefore, in order to function as a signaling messenger, H2O2 does not only need to be produced, but also to be enzymatically degraded or removed. This degradation/removal process is essential to reduce the refractory time, which is defined as the recovery period necessary for detecting successive signals [98]. Otherwise, H2O2 levels will build up, thereby preventing further signaling. Over the years, it has become clear that changes in peroxisomal H2O2 production or CAT activity can modulate the cellular thiol-disulfide state [99]. In the following sections, we first outline what is known about the molecular targets of peroxisome-derived H2O2 and how changes in CAT activity alter cellular thiol-disulfide homeostasis (see Section 4.1). Next, we discuss how peroxisomes may act as modulators of diverse biological processes regulated by H2O2 (see Section 4.2). Finally, we provide a brief overview of how imbalances in any of these processes may contribute to disease (see Section 4.3).
4.1. Molecular Targets
As outlined above (see Section 2), protein cysteinyl residues are the prime mediators of H2O2 signaling [100]. Unfortunately, until now, no global proteomics data are available for redox-active thiols that can be modified by peroxisome-derived H2O2. To address this gap, we recently developed a human HEK-293 cell line that can be used to selectively induce H2O2 production inside peroxisomes in a time- and dose-controlled manner [101], and studies to inventory the peroxisomal H2O2-dependent sulfenome are ongoing. However, during the validation of this cell model, we could provide evidence that peroxisome-derived H2O2 can oxidize redox-sensitive cysteine thiols in proteins within and outside the peroxisomal compartment. Specific examples include the forkhead box O3 transcription factor FOXO3, the p50 and p65 subunits of the transcription factor nuclear factor kappa B (NF-κB), the tumor suppressor phosphatase PTEN, the peroxisomal import receptor PEX5, and the antioxidant enzyme PRDX5 [101].
An alternative approach to study the role of peroxisomal H2O2 metabolism in cellular thiol-disulfide homeostasis is to interfere with CAT activity. Here it is interesting to mention that (i) CAT activity, and not the cellular glutathione levels, appear to dominate the resistance of cells to ROS [102], (ii) treatment of Chang liver cells with 3-amino-1,2,4-triazole, an irreversible inhibitor of CAT activity, increases the protein disulfide levels by 20% [103], and (iii) cardiac-specific overexpression of CAT in mice decreases oxidative cysteine modification of cardiac proteins [104]. Whether the observed changes in cysteine oxidation are caused by alterations in the release of peroxisome-derived H2O2 or by changes in the capacity of peroxisomes to scavenge extra-peroxisomal H2O2 (Figure 2), remains to be determined.
4.2. Biological Processes
Despite the limited number of protein targets that have been identified for peroxisome-derived H2O2 (see Section 4.1), an extensive literature exists on the potential involvement of peroxisomes in the H2O2-mediated regulation of various fundamental biological processes. The aim of this Section is not to provide an exhaustive overview of this subject, but to outline some relevant examples (Figure 3).
4.2.1. Gene Expression
The synthesis, stability, subcellular localization, and/or activity of many transcription factors are regulated by H2O2 [36]. Currently, there is strong evidence that also peroxisomal H2O2 metabolism may contribute to these processes, thereby modulating gene expression. This is perhaps best illustrated by the observation that whole-genome expression profiling studies have revealed that CAT activity modulates the expression of numerous genes, both in human cells [105] and in mice [106]. Another example is that, depending on the cell type and experimental conditions [107], sustained production of peroxisomal H2O2 [108,109,110], CAT inhibition [111], or CAT overexpression [112] can activate [108,109,110] or inhibit [111,112] the activity of NF-κB. These and other findings provide empirical evidence that alterations in peroxisomal H2O2 metabolism can modulate gene expression (Figure 3). However, the biological significance and underlying mechanisms of these observations remain largely to be established.
4.2.2. Cell Fate Regulation
Cell fate decisions such as growth, proliferation, differentiation, senescence, and apoptosis are impacted by multiple environmental and biological cues, including H2O2 [113]. This molecule can instruct such decisions, directly or indirectly, by affecting the functionality of transcription factors and/or other proteins (e.g., kinases, phosphatases, proteases, antioxidant enzymes, etc.) involved in key signal transduction pathways [114]. Importantly, changes in peroxisomal H2O2 metabolism have been reported to influence cell fate transitions [99]. However, although these transition switches can be expected to occur through modification of the epigenetic landscape [115] and transcriptional responses (see Section 4.2.1), the driving mechanisms remain largely to be established.
Given that low levels of H2O2 can promote cell proliferation and differentiation [113], it is not surprising that CAT overexpression has been shown to reduce the growth of various cell types (e.g., rat aortic smooth muscle cells [116], human aortic endothelial cells [117], human MCF-7 breast cancer cells [118], A-375 amelanotic melanoma cells [105], and human promyelocytic HL-60 cells [119]). Interestingly, such treatment has been reported to delay the resting (G0)/gap phase 1 (G1) to synthesis (S)-phase transition in mouse aortic endothelial cells during cell cycle progression [120]. In addition, CAT overexpression in HL-60 (or the human promonocytic cell line U-937) and A-375 cells has been demonstrated to potentiate macrophage and melanocyte differentiation, respectively [105,121].
Depending on the cell type and context, disturbances in peroxisomal H2O2 metabolism may also drive cell transformation (see Section 4.3.5), promote cellular aging and senescence, or trigger cell death. Indeed, chronic reduction of CAT activity in human cells has been documented to increase oxidative damage, enhance the secretion of matrix metalloproteinases, and impair mitochondrial function (see Section 4.2.3) (Figure 3) [122]. In addition, there is strong evidence that CAT can act as a protectant against apoptosis induced by multiple types of oxidative insults (e.g., ultraviolet and ionizing radiation [123,124], arsenic trioxide treatment [125], or P53-induced oxidative stress [126]). Also, peroxisomally located PRDX5 has been shown to have a cytoprotective effect against H2O2-induced cytotoxicity [127]. Importantly, overexpression of CAT may also sensitize cells (e.g., mouse hepatocytes and fibroblasts [128], human alveolar macrophages [129], and MCF-7 cells [118]) as well as animals (e.g., non-obese diabetic mice [130]) to different types of stressors (e.g., paraquat [128], tumor necrosis factor-alpha (TNFα) [128], asbestos [129], cyclophosphamide [130], paclitaxel [118], etoposide [118], and arsenic trioxide [118]). The latter findings clearly demonstrate that high CAT activity can dampen H2O2 signaling. Finally, it has been reported that the lipotoxicity of saturated non-esterified fatty acids in rat insulin-producing cells, which are catalase-poor, is caused by excessive production of H2O2 via peroxisomal β-oxidation [131].
4.2.3. Mitochondrial Function
Peroxisomes and mitochondria cooperate in various metabolic (e.g., β-oxidation of fatty acids, ROS metabolism) and signaling (e.g., antiviral innate immune signaling) pathways [48,132], and disturbances in any of the peroxisomal processes—including H2O2 metabolism—can result in reduced mitochondrial fitness [133,134]. For instance, several studies have documented that inhibition of CAT activity rapidly increases mitochondrial ROS levels [135,136] and impairs mitochondrial membrane potential and aconitase activity [122,137]. In addition, CAT overexpression has been reported to safeguard mitochondrial fitness, thereby protecting the cells against stress insults [122,136,138]. Also here, whether this redox interplay between peroxisomes and mitochondria is a direct result of alterations in the release of peroxisome-derived H2O2 or caused by changes in the capacity of peroxisomes to scavenge extra-peroxisomal H2O2, remains to be investigated. Indeed, to the best of our knowledge, evidence demonstrating that peroxisome-derived H2O2 can directly impact mitochondrial function is still lacking.
4.3. Diseases
Peroxisomes have the intrinsic potential to mediate (see Section 3.1 and Section 3.3) and modulate (see Section 3.2 and Section 3.3) H2O2-driven signaling events. As such, it may not come as a surprise that imbalances in peroxisomal H2O2 metabolism have been associated with multiple oxidative stress-related disease states, including obesity, diabetes, ischemia reperfusion, noise-induced hearing loss, neurodegeneration, aging, and tumor initiation and progression. For a detailed overview on this topic, we refer the reader to another recent review [99]. However, to illustrate the concept, we focus here on some other examples that can all be linked to alterations in CAT activity (Figure 3). Note that the role of this enzyme in protecting cells and tissues against H2O2-induced injury has already been amply documented (see below). In addition, from these and other findings, it is clear that inherited catalase deficiencies should be considered as a strong risk factor for aging-related pathological changes [74].
4.3.1. Heart Disease
Transgenic overexpression of CAT in mouse hearts has been reported (i) to prevent adverse myocardial remodeling and progression to overt heart failure in a mouse model of dilated cardiomyopathy [139], (ii) to protect the heart from (post-)ischemia-reperfusion injury [140,141], diabetic cardiomyopathy and dysfunction [112,142], and lipopolysaccharide-induced cardiac dysfunction and mortality [143], (iii) to preserve cardiac function after myocardial infarction, at least at later time points [144], and (iv) to alleviate cardiac diseases and aging [104,145]. These improved phenotypes have been linked to an overall decrease in oxidative stress [142,143], a shift of the protein thiol/disulfide balance towards thiols [104], a decrease in endothelial nitric oxide synthase activity [142], less nitration of key enzymes involved in energy metabolism [112], a decline in NF-κB signaling [112], a reduction in proinflammatory cytokine release [144], and a marked protection against myocyte hypertrophy, myocyte apoptosis, and interstitial fibrosis [139].
4.3.2. Kidney Disease
CAT overexpression has also been demonstrated to provide renoprotection. Indeed, overexpression of this enzyme in renal proximal tubular cells of angiotensinogen transgenic mice [146], type 1 diabetic Akita transgenic mice [147,148], or type 2 diabetic db/db mice [149] has been shown to mitigate oxidative stress and prevent hypertension, albuminuria, tubulointerstitial fibrosis, and tubular apoptosis [146,147,148]. At the molecular level, these phenomena have been associated with attenuated angiotensinogen and proapoptotic gene expression [149,150].
4.3.3. Insulin Resistance and Diabetes
Pancreatic β-cells are very vulnerable to oxidative stress, a phenomenon of importance in type 1 diabetes and islet transplantation [151]. Interestingly, overexpression of CAT in murine pancreatic β-cells has been demonstrated to have no detrimental effects and to provide marked protection of islet insulin secretion against H2O2- and streptozocin-mediated β-cell dysfunction [152]. In addition, it has been reported that overexpression of CAT improves mitochondrial function in insulin resistant muscle cells, thereby enhancing glucose and fatty acid metabolism [138]. This finding is in line with other studies showing that (i) CAT deletion exacerbates the pre-diabetic phenotype in mice [106], and (ii) overexpression of CAT in obese mice has a positive influence on energy expenditure and metabolic parameters such as leptin and adiponectin levels [153].
4.3.4. Cardiovascular Disease
Oxidative stress is a potent contributing factor to cardiovascular disease [154], and overexpression of CAT in vascular [155,156] or aortic [157] smooth muscle cells has been shown (i) to decrease oxidized lipid-induced cytotoxicity in vitro [155], and (ii) to prevent pathological mechanical changes underlying abdominal aneurysm formation in transgenic mice, primarily through modulation of matrix metalloproteinase activity [156,157]. However, transgenic mice with specific overexpression of CAT in myeloid lineage cells display impaired post-ischemic neovascularization, a phenotype associated with a blunted inflammatory response (e.g., lower levels of inflammatory markers; reduced macrophage infiltration) in ischemic tissues [158].
4.3.5. Cancer
Compared to normal cells, cancer cells frequently produce elevated levels of ROS compared to their normal counterparts [159]. These molecules can act as pro-tumorigenic signals that promote, among others, abnormal cell growth, migration, resistance to apoptosis, adaptations to hypoxia, and genetic instability [159]. Interestingly, low CAT activity has been associated with an increased risk factor for many different cancers, including skin cancer [160], colorectal cancer [161], breast cancer [162], invasive cervical cancer [163], ovarian cancer [164], and prostate cancer [165,166,167]. On the other hand, overexpression of CAT in MCF-7 mammary cancer cells has been reported to result in a less aggressive phenotype and an altered response to chemotherapy [118]. Finally, CAT activity has also been linked to the cathepsin-induced migration and invasion of human lung cancer cells [168].
4.3.6. Neurodegenerative Disease
Oxidative stress is a common denominator of various neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [169]. The underlying reason is that the brain is exquisitely vulnerable to oxidative damage because of its high oxygen consumption, elevated concentrations of unsaturated lipids, and modest antioxidant defense compared to other tissues [169,170]. Interestingly, patients suffering from a peroxisomal deficiency also typically develop severe neurological deficits [171]. However, at the moment, little is known about how alterations in peroxisomal H2O2 metabolism contribute to brain homeostasis and health. To the best of our knowledge, there are no reports that comprehensively study how selective alterations in peroxisomal H2O2 production influence physiological and pathological brain processes. Nonetheless, there are a number of in vitro and in vivo studies that investigate the role of CAT as neuroprotective agent. For example, it has been demonstrated that increased levels of (peroxisome-targeted) CAT (i) protect cultured primary human neurons from H2O2-mediated cytotoxicity [172], (ii) safeguard cultured SH-SY5Y human neuroblastoma cells from β-amyloid-induced oxidative stress [173], (iii) reduce the toxicity of amyloid-β25–35 in rat brain [174], and (iv) protect isolated rat brain mitochondria against the toxic effects of 6-hydroxydopamine on mitochondrial respiration [175]. In addition, there is compelling evidence that binding of the neurotoxic β-amyloid peptide to CAT decreases its activity [176], a phenomenon that may explain why CAT activity is reduced in the brain of Alzheimer’s disease patients [177]. Nevertheless, despite these observations, other studies do not support the idea of CAT being a susceptibility factor for Alzheimer’s disease [178], Parkinson’s disease [179,180], or familial amyotrophic lateral sclerosis [181].
5. Conclusions, Challenges, and Perspectives
As reviewed here, there is currently overwhelming evidence supporting the view that peroxisomes have the intrinsic ability to mediate and modulate H2O2-driven biological processes. In addition, there is growing consensus that perturbations in peroxisomal H2O2 metabolism can elicit adaptive or maladaptive responses that mitigate or aggravate the impact of the underlying cause. However, the specific mechanisms and physiological consequences of these events remain largely to be explored. The most critical questions that need to be answered include (i) the identification and functional dissection of redox-sensitive proteins that can be reversibly oxidized by peroxisome-derived H2O2, (ii) the nature of the proteins involved in the transport of H2O2 and other relevant redox species across the peroxisomal membrane, and (iii) the biological consequences of changes in peroxisomal H2O2 metabolism on cellular signaling networks that drive physiological or pathological responses. These questions are further elaborated in the next sections.
As mentioned above (see Section 4.1), there are presently no mammalian proteomics data for redox-active thiols that can be modified by peroxisome-derived H2O2. The main underlying reason can be attributed to the fact that, until recently, an experimental model to selectively produce physiological concentrations of H2O2 inside peroxisomes in a time- and dose-controlled manner was lacking. However, given that such a model [101] as well as a genetic tool to capture and affinity purify sulfenic acid-containing proteins [182] are currently available, proteome-wide unbiased identification of primary targets of peroxisome-derived H2O2 can be expected soon. In this context, it will also be interesting to see whether H2O2 generated inside peroxisomes or other locations (e.g., mitochondria or the cytosol) yields many specific or common targets.
The steady-state levels of H2O2 inside peroxisomes are determined by the rates of its synthesis, degradation, and diffusion. Besides catalase, also NAD(P)H- and GSH-powered redox systems can be expected to play a role in peroxisomal redox homeostasis (these systems form a complex network of interactions with GPXs, TXNs, and PRDXs) [30,183]. However, as for H2O2 (see Section 3.3), it is still unclear how GSH, GSSG, NAD(P)+, and NAD(P)H are transported across the peroxisomal membrane and how peroxisomes regulate their GSH/GSSG and NAD(P)+/NAD(P)H pools. In addition, the electron donor for peroxisomal PRDX5 remains to be identified. For more details on these topics, we refer the reader to other recent reviews [48,99].
A last unresolved but pertinent question is how alterations in peroxisomal H2O2 metabolism contribute to cellular and organismal physiology. Here, it is important to highlight that more research needs to be done to determine under which conditions peroxisomes serve as net sources or sinks for H2O2. In addition, despite the fact that the redox proteome can provide a link between metabolism and sulfur switch-controlled signaling events [183], it is important to realize that (i) the subgroup of H2O2-sensitive cysteine residues shows less conservation than their redox-insensitive counterparts, and (ii) the H2O2-dependent redoxome can vary dramatically between different cell types [100]. These factors complicate the interpretation of how peroxisome-derived H2O2 may modulate redox-driven intracellular signaling events and pose a significant challenge for translating the in cellulo data to in vivo models and subsequently to clinical practice.
Taken together, there is convincing evidence that peroxisomes do serve as an intracellular hub in H2O2 metabolism and signaling. However, additional work is needed to better understand how cells decode and integrate these cues to produce coherent responses. The outcome of such studies can be expected to advance redox medicine.
Author Contributions
M.F. conceived the format of the manuscript; M.F. and C.L. wrote the manuscript; and C.L., I.R., and M.F. prepared the figures and edited the manuscript.
Funding
M.F. is supported by grants from the KU Leuven (C14/18/088), the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (Onderzoeksprojecten G095315N, G091819N, and GOA8619N), the ERA-Net for Research Programmes on Rare Diseases (PERescue), and H2020-MSCA-ITN (812968). C.L. is supported by the KU Leuven (PDM/18/188).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACOX | Acyl-CoA oxidase |
| Acyl-CoA | Acyl-coenzyme A |
| ABL | Abelson tyrosine-protein kinase |
| CAT | Catalase |
| ERO | Endoplasmic reticulum oxidoreductase |
| FAD | Flavin adenine dinucleotide |
| FMN | Flavin mononucleotide |
| GLRX | Glutaredoxin |
| GPX | Glutathione peroxidase |
| GSH | Glutathione, reduced |
| GSK-3β | Glycogen synthase kinase 3β |
| GSSG | Glutathione, oxidized |
| GSR | Glutathione-disulfide reductase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| NAD(P)+ | Nicotinamide adenine dinucleotide (phosphate), oxidized |
| NAD(P)H | Nicotinamide adenine dinucleotide (phosphate), reduced |
| NFE2L2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | Nuclear factor kappa B |
| NOX | NADPH oxidase |
| PEX | Peroxin |
| PMP | Peroxisomal membrane protein |
| PRDX | Peroxiredoxin |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| TXN | Thioredoxin |
| TXNRD | Thioredoxin reductase |
References
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 2011, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and reactive oxygen species signaling: Interaction of the NADPH oxidase NOX1-5 and superoxide dismutase 1-3 signal transduction pathways. Antioxid. Redox Signal. 2019, 30, 443–486. [Google Scholar] [CrossRef]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A. Peroxisomal disorders: Improved laboratory diagnosis, new defects and the complicated route to treatment. Mol. Cell. Probes 2018, 40, 60–69. [Google Scholar] [CrossRef]
- Wang, B.; Apanasets, O.; Nordgren, M.; Fransen, M. Dissecting peroxisome-mediated signaling pathways: A new and exciting research field. In Molecular Machines Involved in Peroxisome Biogenesis and Maintenance, 1st ed.; Brocard, C., Hartig, A., Eds.; Springer: Wien, Austria, 2014; pp. 255–273. [Google Scholar] [CrossRef]
- Fransen, M. Peroxisome dynamics: Molecular players, mechanisms, and (dys)functions. ISRN Cell Biol. 2012, 2012, 714192. [Google Scholar] [CrossRef]
- Shai, N.; Schuldiner, M.; Zalckvar, E. No peroxisome is an island—Peroxisome contact sites. Biochim. Biophys. Acta 2016, 1863, 1061–1069. [Google Scholar] [CrossRef]
- Distel, B.; Erdmann, R.; Gould, S.J.; Blobel, G.; Crane, D.I.; Cregg, J.M.; Dodt, G.; Fujiki, Y.; Goodman, J.M.; Just, W.W.; et al. A unified nomenclature for peroxisome biogenesis factors. J. Cell Biol. 1996, 135, 1–3. [Google Scholar] [CrossRef]
- Nordgren, M.; Wang, B.; Apanasets, O.; Fransen, M. Peroxisome degradation in mammals: Mechanisms of action, recent advances, and perspectives. Front. Physiol. 2013, 4, 145. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Wong, C.P.; Xu, Z.; Power, C.; Hobman, T.C. Targeted elimination of peroxisomes during viral infection: Lessons from HIV and other viruses. DNA Cell Biol. 2018, 37, 417–421. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65. [Google Scholar] [CrossRef]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef]
- Berridge, M.J. Signaling defects and disease. Cell Signal. Biol. 2014, 12.1–12.66. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Xiao, Z.; La Fontaine, S.; Bush, A.I.; Wedd, A.G. Molecular mechanisms of glutaredoxin enzymes: Versatile hubs for thiol-disulfide exchange between protein thiols and glutathione. J. Mol. Biol. 2019, 431, 158–177. [Google Scholar] [CrossRef]
- Netto, L.E.; Antunes, F. The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, A.; Shi, M.; Chen, X.; Liu, R.; Li, T.; Zhang, C.; Zhang, Z.; Zhu, L.; Ju, Z.; et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018, 13, 2362–2386. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Peroxisomes and cellular oxidant/antioxidant balance: Protein redox modifications and impact on inter-organelle communication. Subcell. Biochem. 2018, 89, 435–461. [Google Scholar] [CrossRef]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Van Rompuy, P.; Fransen, M.; De Béthune, B.; Mannaerts, G.P. Large-scale purification and further characterization of rat pristanoyl-CoA oxidase. Eur. J. Biochem. 1994, 222, 795–801. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Van Rompuy, P.; Vanhooren, J.C.; Mannaerts, G.P. Purification and further characterization of peroxisomal trihydroxycoprostanoyl-CoA oxidase from rat liver. Biochem. J. 1994, 304, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell. Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef]
- Pollegioni, L.; Piubelli, L.; Sacchi, S.; Pilone, M.S.; Molla, G. Physiological functions of D-amino acid oxidases: From yeast to humans. Cell. Mol. Life Sci. 2007, 64, 1373–1394. [Google Scholar] [CrossRef]
- Wu, T.; Yankovskaya, V.; McIntire, W.S. Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 2003, 278, 20514–20525. [Google Scholar] [CrossRef]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Identification and characterization of HAOX1, HAOX2, and HAOX3, three human peroxisomal 2-hydroxy acid oxidases. J. Biol. Chem. 2000, 275, 12590–12597. [Google Scholar] [CrossRef]
- Peduto, A.; Baumgartner, M.R.; Verhoeven, N.M.; Rabier, D.; Spada, M.; Nassogne, M.C.; Poll-The, B.T.; Bonetti, G.; Jakobs, C.; Saudubray, J.M. Hyperpipecolic acidaemia: A diagnostic tool for peroxisomal disorders. Mol. Genet. Metab. 2004, 82, 224–230. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef]
- Yamamoto, K.; Völkl, A.; Fahimi, H.D. Investigation of peroxisomal lipid beta-oxidation enzymes in guinea pig liver peroxisomes by immunoblotting and immunocytochemistry. J. Histochem. Cytochem. 1992, 40, 1909–1918. [Google Scholar] [CrossRef]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef]
- Rafikov, R.; Kumar, S.; Aggarwal, S.; Hou, Y.; Kangath, A.; Pardo, D.; Fineman, J.R.; Black, S. Endothelin-1 stimulates catalase activity through the PKCδ-mediated phosphorylation of serine 167. Free Radic. Biol. Med. 2014, 67, 255–264. [Google Scholar] [CrossRef]
- Cao, C.; Leng, Y.; Kufe, D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J. Biol. Chem. 2003, 278, 29667–29675. [Google Scholar] [CrossRef]
- Ghosh, S.; Janocha, A.J.; Aronica, M.A.; Swaidani, S.; Comhair, S.A.; Xu, W.; Zheng, L.; Kaveti, S.; Kinter, M.; Hazen, S.L.; et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J. Immunol. 2006, 176, 5587–5597. [Google Scholar] [CrossRef]
- Foye, W.O.; Solis, M.C. Inhibition of catalase and lactate dehydrogenase by radiation-protective thiols and thiol derivatives. J. Pharm. Sci. 1969, 58, 352–355. [Google Scholar] [CrossRef]
- Sun, Y.; Oberley, L.W. The inhibition of catalase by glutathione. Free Radic. Biol. Med. 1989, 7, 595–602. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteomics 2014, 96, 253–262. [Google Scholar] [CrossRef]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Geoghegan, V.; Guo, A.; Trudgian, D.; Thomas, B.; Acuto, O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015, 6, 6758. [Google Scholar] [CrossRef]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence inhuman cells. Sci. Signal. 2016, 9, rs9. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Guo, D.; Han, J.; Adam, B.L.; Colburn, N.H.; Wang, M.H.; Dong, Z.; Eizirik, D.L.; She, J.X.; Wang, C.Y. Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochem. Biophys. Res. Commun. 2005, 337, 1308–1318. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.H.; Cheng, W.H.; Bao, Y.; Ho, Y.S.; Reddi, A.R.; Holmgren, A.; Arnér, E.S. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef]
- Góth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age-related disorders? Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Knoops, B.; Argyropoulou, V.; Becker, S.; Ferté, L.; Kuznetsova, O. Multiple Roles of Peroxiredoxins in Inflammation. Mol. Cells 2016, 39, 60–64. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Villa, P.; Eberini, I.; Vandekerckhove, J.; Gianazza, E.; Ghezzi, P. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics 2003, 3, 1154–1161. [Google Scholar] [CrossRef]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef]
- Smeets, A.; Marchand, C.; Linard, D.; Knoops, B.; Declercq, J.P. The crystal structures of oxidized forms of human peroxiredoxin 5 with an intramolecular disulfide bond confirm the proposed enzymatic mechanism for atypical 2-Cys peroxiredoxins. Arch. Biochem. Biophys. 2008, 477, 98–104. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Zamocky, M.; Furtmüller, P.G.; Obinger, C. Evolution of catalases from bacteria to humans. Antioxid. Redox Signal. 2008, 10, 1527–1548. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Singh, A.K.; Gupta, M.K.; Orak, J.K. Antioxidant enzymes in peroxisomes: Effect of ischemia. Ann. N. Y. Acad. Sci. 1996, 804, 696–697. [Google Scholar] [CrossRef]
- Antunes, F.; Cadenas, E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000, 475, 121–126. [Google Scholar] [CrossRef]
- Branco, M.R.; Marinho, H.S.; Cyrne, L.; Antunes, F. Decrease of H2O2 plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 6501–6506. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Mueller, S.; Weber, A.; Fritz, R.; Mütze, S.; Rost, D.; Walczak, H.; Völkl, A.; Stremmel, W. Sensitive and real-time determination of H2O2 release from intact peroxisomes. Biochem. J. 2002, 363, 483–491. [Google Scholar] [CrossRef]
- Li, Y.; Tharappel, J.C.; Cooper, S.; Glenn, M.; Glauert, H.P.; Spear, B.T. Expression of the hydrogen peroxide-generating enzyme fatty acyl CoA oxidase activates NF-kappaB. DNA Cell Biol. 2000, 19, 113–120. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef]
- Rokka, A.; Antonenkov, V.D.; Soininen, R.; Immonen, H.L.; Pirilä, P.L.; Bergmann, U.; Sormunen, R.T.; Weckström, M.; Benz, R.; Hiltunen, J.K. Pxmp2 is a channel-forming protein in mammalian peroxisomal membrane. PLoS ONE 2009, 4, e5090. [Google Scholar] [CrossRef]
- Gualdron-López, M.; Vapola, M.H.; Miinalainen, I.J.; Hiltunen, J.K.; Michels, P.A.; Antonenkov, V.D. Channel-forming activities in the glycosomal fraction from the bloodstream form of Trypanosoma brucei. PLoS ONE 2012, 7, e34530. [Google Scholar] [CrossRef]
- Mindthoff, S.; Grunau, S.; Steinfort, L.L.; Girzalsky, W.; Hiltunen, J.K.; Erdmann, R.; Antonenkov, V.D. Peroxisomal Pex11 is a pore-forming protein homologous to TRPM channels. Biochim. Biophys. Acta 2016, 1863, 271–283. [Google Scholar] [CrossRef]
- Lismont, C.; Koster, J.; Provost, S.; Baes, M.; Van Veldhoven, P.P.; Waterham, H.R.; Fransen, M. Deciphering the potential involvement of PXMP2 and PEX11B in hydrogen peroxide permeation across the peroxisomal membrane reveals a role for PEX11B in protein sorting. Biochim. Biophys. Acta Biomembr. 2019. [Google Scholar] [CrossRef]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef]
- Vestergaard, C.L.; Flyvbjerg, H.; Møller, I.M. Intracellular signaling by diffusion: Can waves of hydrogen peroxide transmit intracellular information in plant cells? Front. Plant Sci. 2012, 3, 295. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Redox signaling from and to peroxisomes: Progress, challenges, and prospects. Antioxid. Redox Signal. 2019, 30, 95–112. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; Sun, M.; Tian, C.; Sun, R.; Morales Betanzos, C.; Tallman, K.A.; Porter, N.A.; Yang, Y.; Guo, D.; et al. Systematic and quantitative assessment of hydrogen peroxide reactivity with cysteines across human proteomes. Mol. Cell. Proteom. 2017, 16, 1815–1828. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Brees, C.; Knoops, B.; Van Veldhoven, P.P.; Fransen, M. Peroxisomes as modulators of cellular protein thiol oxidation: A new model system. Antioxid. Redox Signal. 2019, 30, 22–39. [Google Scholar] [CrossRef]
- Zhao, M.X.; Wen, J.L.; Wang, L.; Wang, X.P.; Chen, T.S. Intracellular catalase activity instead of glutathione level dominates the resistance of cells to reactive oxygen species. Cell Stress Chaperones 2019, 24, 609–619. [Google Scholar] [CrossRef]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Behring, J.B.; Shao, D.; Sverdlov, A.L.; Whelan, S.A.; Elezaby, A.; Yin, X.; Siwik, D.A.; Seta, F.; Costello, C.E.; et al. Overexpression of catalase diminishes oxidative cysteine modifications of cardiac proteins. PLoS ONE 2015, 10, e0144025. [Google Scholar] [CrossRef]
- Bracalente, C.; Ibañez, I.L.; Berenstein, A.; Notcovich, C.; Cerda, M.B.; Klamt, F.; Chernomoretz, A.; Durán, H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Upregulation of antioxidant genes correlates with regression of melanoma malignancy and with malignant progression when downregulated. Oncotarget 2016, 7, 41154–41171. [Google Scholar] [CrossRef] [Green Version]
- Heit, C.; Marshall, S.; Singh, S.; Yu, X.; Charkoftaki, G.; Zhao, H.; Orlicky, D.J.; Fritz, K.S.; Thompson, D.C.; Vasiliou, V. Catalase deletion promotes prediabetic phenotype in mice. Free Radic. Biol. Med. 2017, 103, 48–56. [Google Scholar] [CrossRef]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Modulation of NF-kappaB-dependent gene expression by H2O2: A major role for a simple chemical process in a complex biological response. Antioxid. Redox Signal. 2009, 11, 2043–2053. [Google Scholar] [CrossRef]
- Chu, R.; Lin, Y.; Reddy, K.C.; Pan, J.; Rao, M.S.; Reddy, J.K.; Yeldandi, A.V. Transformation of epithelial cells stably transfected with H2O2-generating peroxisomal urate oxidase. Cancer Res. 1996, 56, 4846–4852. [Google Scholar]
- Okamoto, M.; Reddy, J.K.; Oyasu, R. Tumorigenic conversion of a non-tumorigenic rat urothelial cell line by overexpression of H2O2-generating peroxisomal fatty acyl-CoA oxidase. Int. J. Cancer 1997, 70, 716–721. [Google Scholar] [CrossRef]
- Dadras, S.S.; Thorgeirsson, S.S.; Rao, M.S.; Reddy, J.K. Implication of hydrogen peroxide generation and apoptosis in the neoplastic transformation of mouse fibroblasts overexpressing peroxisomal fatty acyl-CoA oxidase. Int. J. Oncol. 1998, 12, 37–44. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Lorne, E.; Zhao, X.; Tsuruta, Y.; Sha, Y.; Liu, G.; Abraham, E. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 179, 694–704. [Google Scholar] [CrossRef]
- Cong, W.; Ruan, D.; Xuan, Y.; Niu, C.; Tao, Y.; Wang, Y.; Zhan, K.; Cai, L.; Jin, L.; Tan, Y. Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-κB signaling activation in the heart. J. Mol. Cell. Cardiol. 2015, 89, 314–325. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Tatapudy, S.; Aloisio, F.; Barber, D.; Nystul, T. Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 2017, 18, 2105–2118. [Google Scholar] [CrossRef]
- Farr, R.L.; Lismont, C.; Terlecky, S.R.; Fransen, M. Peroxisome biogenesis in mammalian cells: The impact of genes and environment. Biochim. Biophys. Acta 2016, 1863, 1049–1060. [Google Scholar] [CrossRef]
- Brown, M.R.; Miller, F.J., Jr.; Li, W.G.; Ellingson, A.N.; Mozena, J.D.; Chatterjee, P.; Engelhardt, J.F.; Zwacka, R.M.; Oberley, L.W.; Fang, X.; et al. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ. Res. 1999, 85, 524–533. [Google Scholar] [CrossRef]
- Zanetti, M.; Katusic, Z.S.; O’Brien, T. Adenoviral-mediated overexpression of catalase inhibits endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2620–H2626. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef]
- Hachiya, M.; Akashi, M. Catalase regulates cell growth in HL60 human promyelocytic cells: Evidence for growth regulation by H2O2. Radiat. Res. 2005, 163, 271–282. [Google Scholar] [CrossRef]
- Onumah, O.E.; Jules, G.E.; Zhao, Y.; Zhou, L.; Yang, H.; Guo, Z. Overexpression of catalase delays G0/G1- to S-phase transition during cell cycle progression in mouse aortic endothelial cells. Free Radic. Biol. Med. 2009, 46, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Sakaguchi, N.; Hachiya, M.; Nakayama, F.; Yamakawa, M.; Akashi, M. Role of catalase in monocytic differentiation of U937 cells by TPA: Hydrogen peroxide as a second messenger. Leukemia 2009, 23, 761–769. [Google Scholar] [CrossRef]
- Koepke, J.I.; Wood, C.S.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Progeric effects of catalase inactivation in human cells. Toxicol. Appl. Pharmacol. 2008, 232, 99–108. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Cario-André, M.; Pain, C.; Ged, C.; deVerneuil, H.; Taïeb, A. Protection of normal human reconstructed epidermis from UV by catalase overexpression. Cancer Gene Ther. 2007, 14, 174–186. [Google Scholar] [CrossRef]
- Xiao, X.; Luo, H.; Vanek, K.N.; LaRue, A.C.; Schulte, B.A.; Wang, G.Y. Catalase inhibits ionizing radiation-induced apoptosis in hematopoietic stem and progenitor cells. Stem Cells Dev. 2015, 24, 1342–1351. [Google Scholar] [CrossRef]
- Song, L.L.; Tu, Y.Y.; Xia, L.; Wang, W.W.; Wei, W.; Ma, C.M.; Wen, D.H.; Lei, H.; Xu, H.Z.; Wu, Y.L. Targeting catalase but not peroxiredoxins enhances arsenic trioxide-induced apoptosis in K562 cells. PLoS ONE 2014, 9, e104985. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Catalase protects HepG2 cells from apoptosis induced by DNA-damaging agents by accelerating the degradation of p53. J. Biol. Chem. 2003, 278, 4660–4667. [Google Scholar] [CrossRef]
- Walbrecq, G.; Wang, B.; Becker, S.; Hannotiau, A.; Fransen, M.; Knoops, B. Antioxidant cytoprotection by peroxisomal peroxiredoxin-5. Free Radic. Biol. Med. 2015, 84, 215–226. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Van Remmen, H.; Vijg, J.; Richardson, A. Catalase transgenic mice: Characterization and sensitivity to oxidative stress. Arch. Biochem. Biophys. 2004, 422, 197–210. [Google Scholar] [CrossRef]
- Carter, A.B.; Tephly, L.A.; Venkataraman, S.; Oberley, L.W.; Zhang, Y.; Buettner, G.R.; Spitz, D.R.; Hunninghake, G.W. High levels of catalase and glutathione peroxidase activity dampen H2O2 signaling in human alveolar macrophages. Am. J. Respir. Cell. Mol. Biol. 2004, 31, 43–53. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Epstein, P.N. Metallothionein and catalase sensitize to diabetes in non-obese diabetic mice: Reactive oxygen species may have a protective role in pancreatic beta-cells. Diabetes 2006, 55, 1592–1604. [Google Scholar] [CrossRef]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef]
- Schrader, M.; Kamoshita, M.; Islinger, M. Organelle interplay-peroxisome interactions in health and disease. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef]
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.R.; Sampaio, I.H.; Teodoro, B.G.; Sousa, T.A.; Zoppi, C.C.; Queiroz, A.L.; Passos, M.A.; Alberici, L.C.; Teixeira, F.R.; Manfiolli, A.O.; et al. Hydrogen peroxide production regulates the mitochondrial function in insulin resistant muscle cells: Effect of catalase overexpression. Biochim. Biophys. Acta 2013, 1832, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Lennon-Edwards, S.; Lancel, S.; Biolo, A.; Siwik, D.A.; Pimentel, D.R.; Dorn, G.W.; Kang, Y.J.; Colucci, W.S. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ. Heart Fail. 2010, 3, 306–313. [Google Scholar] [CrossRef]
- Li, G.; Chen, Y.; Saari, J.T.; Kang, Y.J. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Physiol. 1997, 273, H1090–H1095. [Google Scholar] [CrossRef]
- Wölkart, G.; Kaber, G.; Kojda, G.; Brunner, F. Role of endogenous hydrogen peroxide in cardiovascular ischaemia/reperfusion function: Studies in mouse hearts with catalase-overexpression in the vascular endothelium. Pharmacol. Res. 2006, 54, 50–56. [Google Scholar] [CrossRef]
- Dong, F.; Fang, C.X.; Yang, X.; Zhang, X.; Lopez, F.L.; Ren, J. Cardiac overexpression of catalase rescues cardiac contractile dysfunction induced by insulin resistance: Role of oxidative stress, protein carbonyl formation and insulin sensitivity. Diabetologia 2006, 49, 1421–1433. [Google Scholar] [CrossRef]
- Turdi, S.; Han, X.; Huff, A.F.; Roe, N.D.; Hu, N.; Gao, F.; Ren, J. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: Role of autophagy. Free Radic. Biol. Med. 2012, 53, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Pendergrass, K.D.; Varghese, S.T.; Maiellaro-Rafferty, K.; Brown, M.E.; Taylor, W.R.; Davis, M.E. Temporal effects of catalase overexpression on healing after myocardial infarction. Circ. Heart Fail. 2011, 4, 98–106. [Google Scholar] [CrossRef]
- Ren, J.; Li, Q.; Wu, S.; Li, S.Y.; Babcock, S.A. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mech. Ageing Dev. 2007, 128, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.L.; Chénier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lo, C.S.; Chenier, I.; Maachi, H.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Overexpression of catalase prevents hypertension and tubulointerstitial fibrosis and normalization of renal angiotensin-converting enzyme-2 expression in Akita mice. Am. J. Physiol. Renal Physiol. 2013, 304, F1335–F1346. [Google Scholar] [CrossRef]
- Abdo, S.; Shi, Y.; Otoukesh, A.; Ghosh, A.; Lo, C.S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase overexpression prevents nuclear factor erythroid 2-related factor 2 stimulation of renal angiotensinogen gene expression, hypertension, and kidney injury in diabetic mice. Diabetes 2014, 63, 3483–3496. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Chénier, I.; Godin, N.; Zhang, S.L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 2008, 57, 451–459. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Tran, S.; Sachetelli, S.; Zhang, S.L.; Guo, D.F.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int. 2007, 71, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, A.; Kirkwood, T.B.; Shanley, D.P. Metabolic evolution suggests an explanation for the weakness of antioxidant defences in beta-cells. Mech. Ageing Dev. 2009, 130, 216–221. [Google Scholar] [CrossRef]
- Xu, B.; Moritz, J.T.; Epstein, P.N. Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic. Biol. Med. 1999, 27, 830–837. [Google Scholar] [CrossRef]
- Amos, D.L.; Robinson, T.; Massie, M.B.; Cook, C.; Hoffsted, A.; Crain, C.; Santanam, N. Catalase overexpression modulates metabolic parameters in a new ‘stress-less’ leptin-deficient mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2293–2306. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and oxidative stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]
- Santanam, N.; Aug, N.; Zhou, M.; Keshava, C.; Parthasarathy, S. Overexpression of human catalase gene decreases oxidized lipid-induced cytotoxicity in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1912–1917. [Google Scholar] [CrossRef]
- Parastatidis, I.; Weiss, D.; Joseph, G.; Taylor, W.R. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2389–2396. [Google Scholar] [CrossRef]
- Maiellaro-Rafferty, K.; Weiss, D.; Joseph, G.; Wan, W.; Gleason, R.L.; Taylor, W.R. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H355–H362. [Google Scholar] [CrossRef] [Green Version]
- Hodara, R.; Weiss, D.; Joseph, G.; Velasquez-Castano, J.C.; Landázuri, N.; Han, J.W.; Yoon, Y.S.; Taylor, W.R. Overexpression of catalase in myeloid cells causes impaired postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2203–2209. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signaling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Syed, D.N.; Mukhtar, H. Gender bias in skin cancer: Role of catalase revealed. J. Investig. Dermatol. 2012, 132, 512–514. [Google Scholar] [CrossRef]
- Chang, D.; Hu, Z.L.; Zhang, L.; Zhao, Y.S.; Meng, Q.H.; Guan, Q.B.; Zhou, J.; Pan, H.Z. Association of catalase genotype with oxidative stress in the predication of colorectal cancer: Modification by epidemiological factors. Biomed. Environ. Sci. 2012, 25, 156–162. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Chaudhuri, G. Maintenance of higher H2O2 levels, and its mechanism of action to induce growth in breast cancer cells: Important roles of bioactive catalase and PP2A. Free Radic. Biol. Med. 2012, 53, 1541–1551. [Google Scholar] [CrossRef]
- Castaldo, S.A.; da Silva, A.P.; Matos, A.; Inácio, Â.; Bicho, M.; Medeiros, R.; Alho, I.; Bicho, M.C. The role of CYBA (p22phox) and catalase genetic polymorphisms and their possible epistatic interaction in cervical cancer. Tumour Biol. 2015, 36, 909–914. [Google Scholar] [CrossRef]
- Belotte, J.; Fletcher, N.M.; Saed, M.G.; Abusamaan, M.S.; Dyson, G.; Diamond, M.P.; Saed, G.M. A single nucleotide polymorphism in catalase is strongly associated with ovarian cancer survival. PLoS ONE 2015, 10, e0135739. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.; Tian, P.; Shen, K.; Zhu, J.; Feng, M.; Wan, C.; Yang, T.; Chen, L.; Wen, F. The catalase C-262T gene polymorphism and cancer risk: A systematic review and meta-analysis. Medicine 2015, 94, e679. [Google Scholar] [CrossRef]
- Hu, J.; Feng, F.; Zhu, S.; Sun, L.; Li, G.; Jiang, N.; Shang, Z.; Niu, Y. Catalase C-262T polymorphism and risk of prostate cancer: Evidence from meta-analysis. Gene 2015, 558, 265–270. [Google Scholar] [CrossRef]
- Liu, K.; Liu, X.; Wang, M.; Wang, X.; Kang, H.; Lin, S.; Yang, P.; Dai, C.; Xu, P.; Li, S.; et al. Two common functional catalase gene polymorphisms (rs1001179 and rs794316) and cancer susceptibility: Evidence from 14,942 cancer cases and 43,285 controls. Oncotarget 2016, 7, 62954–62965. [Google Scholar] [CrossRef]
- Tsai, J.Y.; Lee, M.J.; Dah-Tsyr Chang, M.; Huang, H. The effect of catalase on migration and invasion of lung cancer cells by regulating the activities of cathepsin S, L, and K. Exp. Cell Res. 2014, 323, 28–40. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in brain development and function. Biochim. Biophys. Acta 2016, 1863, 934–955. [Google Scholar] [CrossRef]
- Singhal, A.; Morris, V.B.; Labhasetwar, V.; Ghorpade, A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013, 4, e903. [Google Scholar] [CrossRef]
- Chilumuri, A.; Odell, M.; Milton, N.G. Benzothiazole aniline tetra(ethylene glycol) and 3-amino-1,2,4-triazole inhibit neuroprotection against amyloid peptides by catalase overexpression in vitro. ACS Chem. Neurosci. 2013, 4, 1501–1512. [Google Scholar] [CrossRef]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted antioxidant, catalase-SKL, reduces beta-amyloid toxicity in the rat brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef]
- Iglesias-González, J.; Sánchez-Iglesias, S.; Méndez-Álvarez, E.; Rose, S.; Hikima, A.; Jenner, P.; Soto-Otero, R. Differential toxicity of 6-hydroxydopamine in SH-SY5Y human neuroblastoma cells and rat brain mitochondria: Protective role of catalase and superoxide dismutase. Neurochem. Res. 2012, 37, 2150–2160. [Google Scholar] [CrossRef]
- Habib, L.K.; Lee, M.T.; Yang, J. Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar] [CrossRef]
- Gsell, W.; Conrad, R.; Hickethier, M.; Sofic, E.; Frölich, L.; Wichart, I.; Jellinger, K.; Moll, G.; Ransmayr, G.; Beckmann, H.; et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of Alzheimer type. J. Neurochem. 1995, 64, 1216–1223. [Google Scholar] [CrossRef]
- Capurso, C.; Solfrizzi, V.; D’Introno, A.; Colacicco, A.M.; Capurso, S.A.; Bifaro, L.; Menga, R.; Santamato, A.; Seripa, D.; Pilotto, A.; et al. Short arm of chromosome 11 and sporadic Alzheimer’s disease: Catalase and cathepsin D gene polymorphisms. Neurosci. Lett. 2008, 432, 237–242. [Google Scholar] [CrossRef]
- Gasser, T.; Wszolek, Z.K.; Trofatter, J.; Ozelius, L.; Uitti, R.J.; Lee, C.S.; Gusella, J.; Pfeiffer, R.F.; Calne, D.B.; Breakefield, X.O. Genetic linkage studies in autosomal dominant parkinsonism: Evaluation of seven candidate genes. Ann. Neurol. 1994, 36, 387–396. [Google Scholar] [CrossRef]
- Parboosingh, J.S.; Rousseau, M.; Rogan, F.; Amit, Z.; Chertkow, H.; Johnson, W.G.; Manganaro, F.; Schipper, H.N.; Curran, T.J.; Stoessl, J.; et al. Absence of mutations in superoxide dismutase and catalase genes in patients with Parkinson’s disease. Arch. Neurol. 1995, 52, 1160–1163. [Google Scholar] [CrossRef]
- Przedborski, S.; Donaldson, D.M.; Murphy, P.L.; Hirsch, O.; Lange, D.; Naini, A.B.; McKenna-Yasek, D.; Brown, R.H., Jr. Blood superoxide dismutase, catalase and glutathione peroxidase activities in familial and sporadic amyotrophic lateral sclerosis. Neurodegeneration 1996, 5, 57–64. [Google Scholar] [CrossRef]
- Waszczak, C.; Akter, S.; Eeckhout, D.; Persiau, G.; Wahni, K.; Bodra, N.; Van Molle, I.; De Smet, B.; Vertommen, D.; Gevaert, K.; et al. Sulfenome mining in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2014, 111, 11545–11550. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The redox code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef]
Figure 1.
General principles of H2O2 signaling. GLRX, glutaredoxin; GSH, reduced glutathione; GSR, glutathione-disulfide reductase; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); TXN, thioredoxin; TXNRD, thioredoxin reductase.
Figure 1.
General principles of H2O2 signaling. GLRX, glutaredoxin; GSH, reduced glutathione; GSR, glutathione-disulfide reductase; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); TXN, thioredoxin; TXNRD, thioredoxin reductase.

Figure 2.
Peroxisomal hydrogen peroxide (H2O2) metabolism and its potential effects on intracellular H2O2 gradients. H2O2-producing and -degrading enzymes are depicted as red and green rectangles, respectively.
Figure 2.
Peroxisomal hydrogen peroxide (H2O2) metabolism and its potential effects on intracellular H2O2 gradients. H2O2-producing and -degrading enzymes are depicted as red and green rectangles, respectively.

Figure 3.
Schematic presentation of how alterations in catalase (CAT) activity may shape intracellular H2O2 gradients, thereby impacting multiple biological processes and contributing to disease initiation and progression. MT, mitochondria.
Figure 3.
Schematic presentation of how alterations in catalase (CAT) activity may shape intracellular H2O2 gradients, thereby impacting multiple biological processes and contributing to disease initiation and progression. MT, mitochondria.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. https://doi.org/10.3390/ijms20153673
AMA Style
Lismont C, Revenco I, Fransen M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. International Journal of Molecular Sciences. 2019; 20(15):3673. https://doi.org/10.3390/ijms20153673
Chicago/Turabian StyleLismont, Celien, Iulia Revenco, and Marc Fransen. 2019. "Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease" International Journal of Molecular Sciences 20, no. 15: 3673. https://doi.org/10.3390/ijms20153673
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.