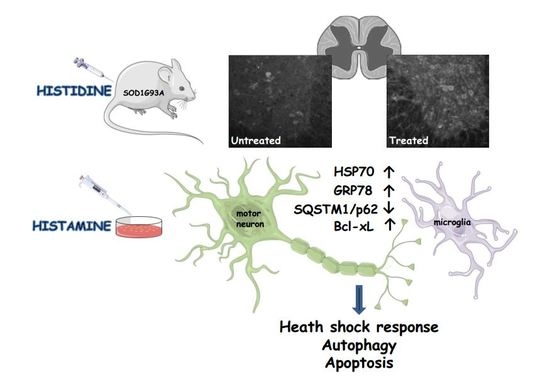
Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Histamine Modulates the Hsps Response in SOD1-G93A Primary Microglia
2.2. Histamine Modulates the Hsps Response in NSC-G93A Motor Neuron Cells
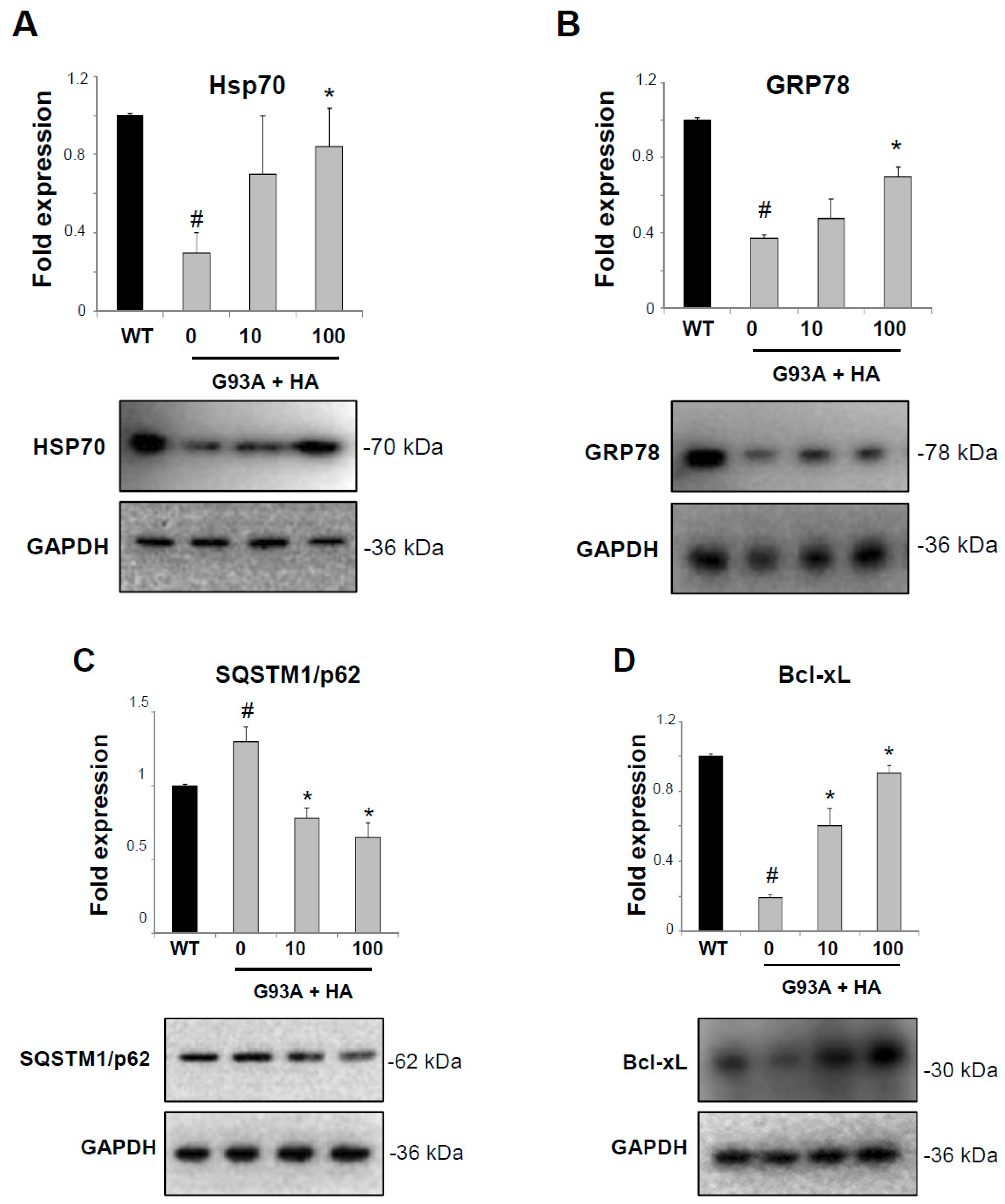
2.3. Histaminergic Signaling Activates the Hsps Response in Spinal Cord from SOD1-G93A Symptomatic Mice
2.4. Histaminergic Signaling Activates the Hsps Response and Prevents Dendritic Spine Loss in Motor Cortex from SOD1-G93A Symptomatic Mice
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Mice
4.3. Primary Microglia Cultures
4.4. Differentiated NSC-34 Motor Neurons
4.5. Spinal Cord and Cortical Tissue Analysis
4.6. Protein Extraction, SDS-PAGE and Western Blotting
4.7. Immunofluorescence and Confocal Microscopy
4.8. Golgi Staining of Cortical Motor Neurons and Measurement of Dendritic Spine Density
4.9. Data Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CNS | Central nervous system |
| SOD1 | Superoxide dismutase 1 |
| HSR | Heat shock response |
| Hsps | Heat shock proteins |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| WT | Wild-type |
References
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Appel, S.H.; Zhao, W.; Beers, D.R.; Henkel, J.S. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011, 30, 4–8. [Google Scholar] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, J.Y.; Ma, H.; Lee, J.E.; Yenari, M.A. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J. Cereb. Blood Flow Metab. 2008, 28, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Lu, C.H.; Greensmith, L. The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol. Ther. 2014, 141, 40–54. [Google Scholar] [CrossRef] [PubMed]
- San Gil, R.; Ooi, L.; Yerbury, J.J.; Ecroyd, H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Atassi, N.; David, W.; Cudkowicz, M.; Schoenfeld, D. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 2018, 90, e565–e574. [Google Scholar] [CrossRef]
- Vukosavic, S.; Dubois-Dauphin, M.; Romero, N.; Przedborski, S. Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1999, 73, 2460–2468. [Google Scholar] [CrossRef]
- Kalmar, B.; Greensmith, L. Cellular Chaperones as Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar] [CrossRef]
- Nassif, M.; Valenzuela, V.; Rojas-Rivera, D.; Vidal, R.; Matus, S.; Castillo, K.; Fuentealba, Y.; Kroemer, G.; Levine, B.; Hetz, C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 2014, 10, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chen, Z. The roles of histamine and its receptor ligands in central nervous system disorders: An update. Pharmacol. Ther. 2017, 175, 116–132. [Google Scholar] [CrossRef]
- Shan, L.; Bao, A.M.; Swaab, D.F. The human histaminergic system in neuropsychiatric disorders. Trends Neurosci. 2015, 38, 167–177. [Google Scholar] [CrossRef]
- Zlomuzica, A.; Dere, D.; Binder, S.; De Souza Silva, M.A.; Huston, J.P.; Dere, E. Neuronal histamine and cognitive symptoms in Alzheimer’s disease. Neuropharmacology 2016, 106, 135–145. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C.; Fernandez-Novoa, L.; Aliev, G. Neuroimmune Crosstalk in CNS Disorders: The Histamine Connection. Curr. Pharm. Des. 2016, 22, 819–848. [Google Scholar] [CrossRef]
- Apolloni, S.; Amadio, S.; Fabbrizio, P.; Morello, G.; Spampinato, A.G.; Latagliata, E.C.; Salvatori, I.; Proietti, D.; Ferri, A.; Madaro, L.; et al. Histaminergic transmission slows progression of amyotrophic lateral sclerosis. J. Cachexia Sarcopenia Muscle 2019. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Verdile, V.; Morello, G.; Iemmolo, R.; Aronica, E.; Cavallaro, S.; Volonte, C. Histamine Regulates the Inflammatory Profile of SOD1-G93A Microglia and the Histaminergic System Is Dysregulated in Amyotrophic Lateral Sclerosis. Front. Immunol. 2017, 8, 1689. [Google Scholar] [CrossRef]
- Clarke, B.E.; Gil, R.S.; Yip, J.; Kalmar, B.; Greensmith, L. Regional differences in the inflammatory and heat shock response in glia: Implications for ALS. Cell Stress Chaperones 2019. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Fabbrizio, P.; Amadio, S.; Apolloni, S.; Volonte, C. P2X7 Receptor Activation Modulates Autophagy in SOD1-G93A Mouse Microglia. Front. Cell. Neurosci. 2017, 11, 249. [Google Scholar] [CrossRef]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell. Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Kalmar, B.; Novoselov, S.; Gray, A.; Cheetham, M.E.; Margulis, B.; Greensmith, L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J. Neurochem. 2008, 107, 339–350. [Google Scholar] [CrossRef]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 2016, 26, 1512–1528. [Google Scholar] [CrossRef]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Mu, E.W.; Noakes, P.G.; Lavidis, N.A.; Bellingham, M.C. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2016, 4, 77. [Google Scholar] [CrossRef]
- Spalloni, A.; Origlia, N.; Sgobio, C.; Trabalza, A.; Nutini, M.; Berretta, N.; Bernardi, G.; Domenici, L.; Ammassari-Teule, M.; Longone, P. Postsynaptic alteration of NR2A subunit and defective autophosphorylation of alphaCaMKII at threonine-286 contribute to abnormal plasticity and morphology of upper motor neurons in presymptomatic SOD1G93A mice, a murine model for amyotrophic lateral sclerosis. Cereb. Cortex 2011, 21, 796–805. [Google Scholar]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Chen, H.J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The heat shock response plays an important role in TDP-43 clearance: Evidence for dysfunction in amyotrophic lateral sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Murshid, A. Molecular Chaperone Accumulation in Cancer and Decrease in Alzheimer’s Disease: The Potential Roles of HSF1. Front. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 2018, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, D.; Nakamura, A.; Hineno, A.; Kobayashi, C.; Kinoshita, T.; Yoshida, K.; Ikeda, S. Elevation of serum heat-shock protein levels in amyotrophic lateral sclerosis. Neurol. Sci. 2016, 37, 1277–1281. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Petri, S.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 246–254. [Google Scholar] [CrossRef]
- Kieran, D.; Kalmar, B.; Dick, J.R.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405. [Google Scholar] [CrossRef]
- Lin, P.Y.; Simon, S.M.; Koh, W.K.; Folorunso, O.; Umbaugh, C.S.; Pierce, A. Heat shock factor 1 over-expression protects against exposure of hydrophobic residues on mutant SOD1 and early mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2013, 8, 43. [Google Scholar] [CrossRef]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.L.; Kilty, R.; Zhang, F.; Zhu, H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 11068–11077. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonte, C. Actions of the antihistaminergic clemastine on presymptomatic SOD1-G93A mice ameliorate ALS disease progression. J. Neuroinflammation 2016, 13, 191. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Kasza, A.; Hunya, A.; Frank, Z.; Fulop, F.; Torok, Z.; Balogh, G.; Santha, M.; Balind, A.; Bernath, S.; Blundell, K.L.; et al. Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 557–571. [Google Scholar] [CrossRef]
- Roe, M.S.; Wahab, B.; Torok, Z.; Horvath, I.; Vigh, L.; Prodromou, C. Dihydropyridines Allosterically Modulate Hsp90 Providing a Novel Mechanism for Heat Shock Protein Co-induction and Neuroprotection. Front. Mol. Biosci. 2018, 5, 51. [Google Scholar] [CrossRef]
- Volonte, C.; Apolloni, S.; Sabatelli, M. Histamine beyond its effects on allergy: Potential therapeutic benefits for the treatment of Amyotrophic Lateral Sclerosis (ALS). Pharmacol. Ther. 2019. [Google Scholar] [CrossRef]
- Apolloni, S.; Parisi, C.; Pesaresi, M.G.; Rossi, S.; Carri, M.T.; Cozzolino, M.; Volonte, C.; D’Ambrosi, N. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J. Immunol. 2013, 190, 5187–5195. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Gibb, R.; Kolb, B. A method for vibratome sectioning of Golgi-Cox stained whole rat brain. J. Neurosci. Methods 1998, 79, 1–4. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apolloni, S.; Caputi, F.; Pignataro, A.; Amadio, S.; Fabbrizio, P.; Ammassari-Teule, M.; Volonté, C. Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS. Int. J. Mol. Sci. 2019, 20, 3793. https://doi.org/10.3390/ijms20153793
Apolloni S, Caputi F, Pignataro A, Amadio S, Fabbrizio P, Ammassari-Teule M, Volonté C. Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS. International Journal of Molecular Sciences. 2019; 20(15):3793. https://doi.org/10.3390/ijms20153793
Chicago/Turabian StyleApolloni, Savina, Francesca Caputi, Annabella Pignataro, Susanna Amadio, Paola Fabbrizio, Martine Ammassari-Teule, and Cinzia Volonté. 2019. "Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS" International Journal of Molecular Sciences 20, no. 15: 3793. https://doi.org/10.3390/ijms20153793