Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities

 ,
,  and
and
Abstract
:
1. Introduction
- (1)
- (2)
- In consequence, every molecule involved in living systems interacts more or less specifically, strongly, and transiently with several others to form the chemical basis of an “interactome” [70].
- (3)
- (4)
- (5)
- Receptors for molecularly complementary ligands evolve complementary functions. For example, a homocomplementary molecule may evolve into a ligand–receptor pair, or heterocomplementary molecules may evolve so that one becomes the ligand and the other the receptor.
- (6)
- As a result of principles 1–5, compounds that are molecularly complementary will alter each other’s physiological activity, and conversely, when compounds alter each other’s physiological activity, they will be found to be molecularly complementary [77].
- (7)
2. Results
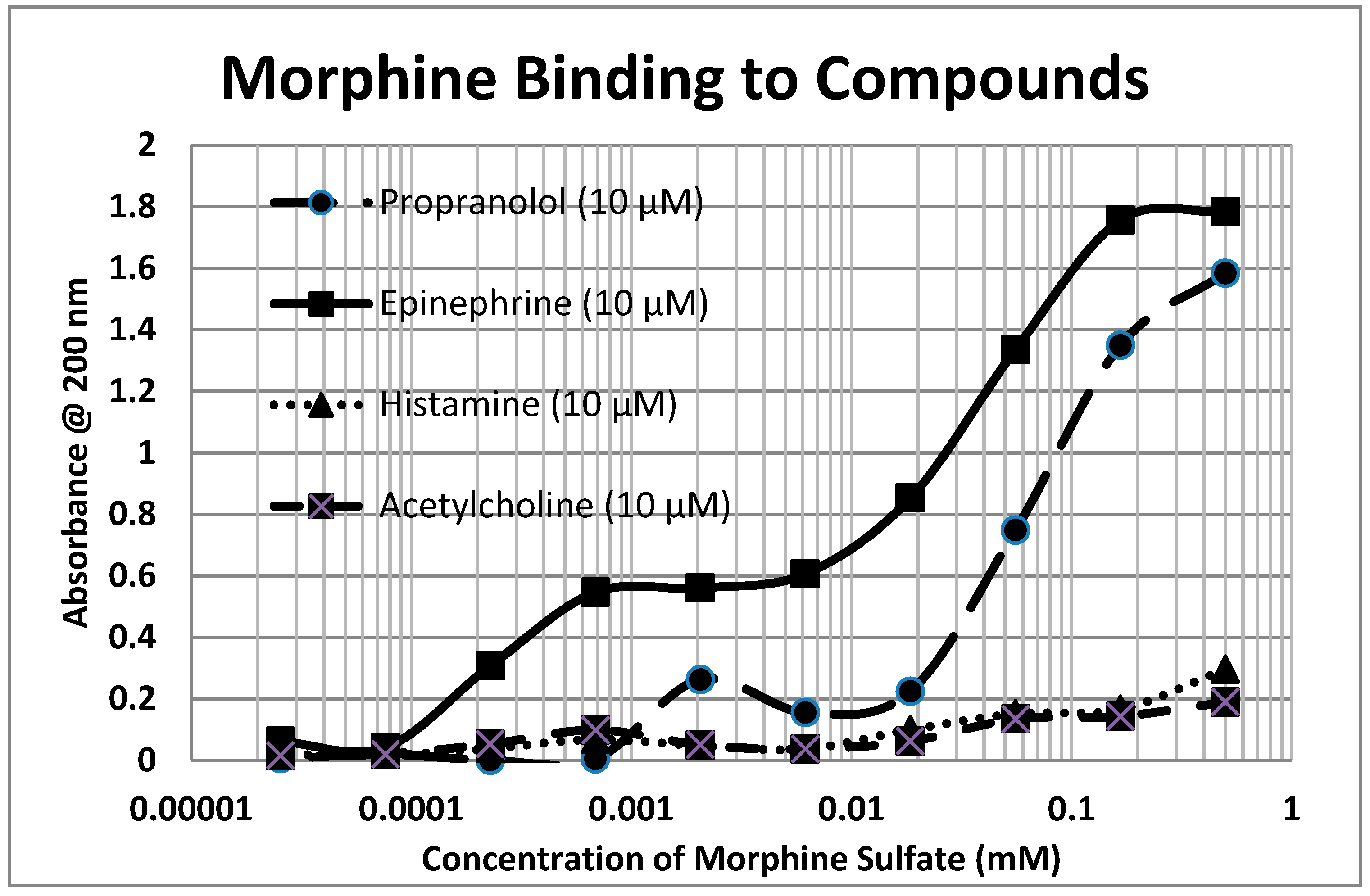
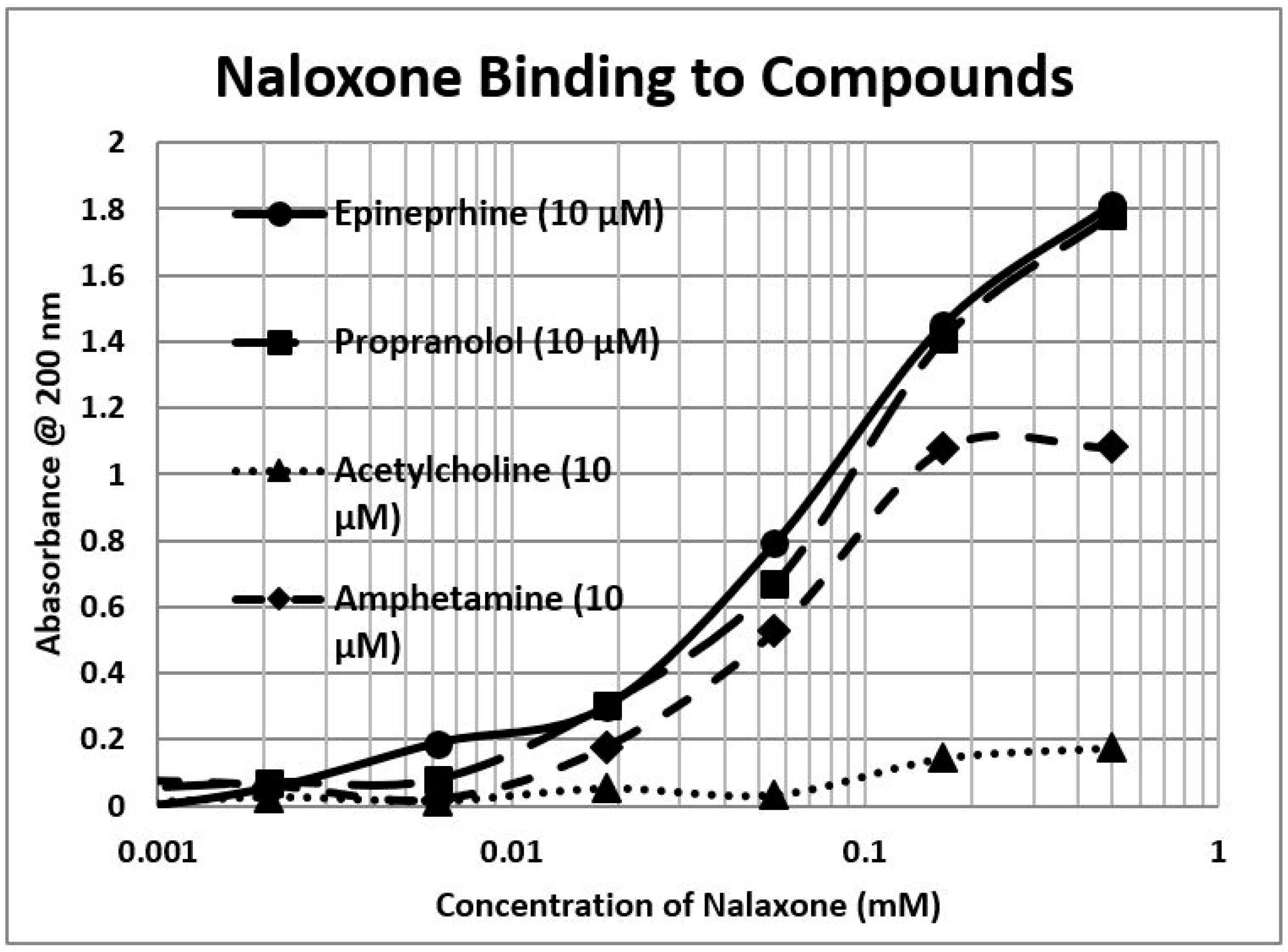
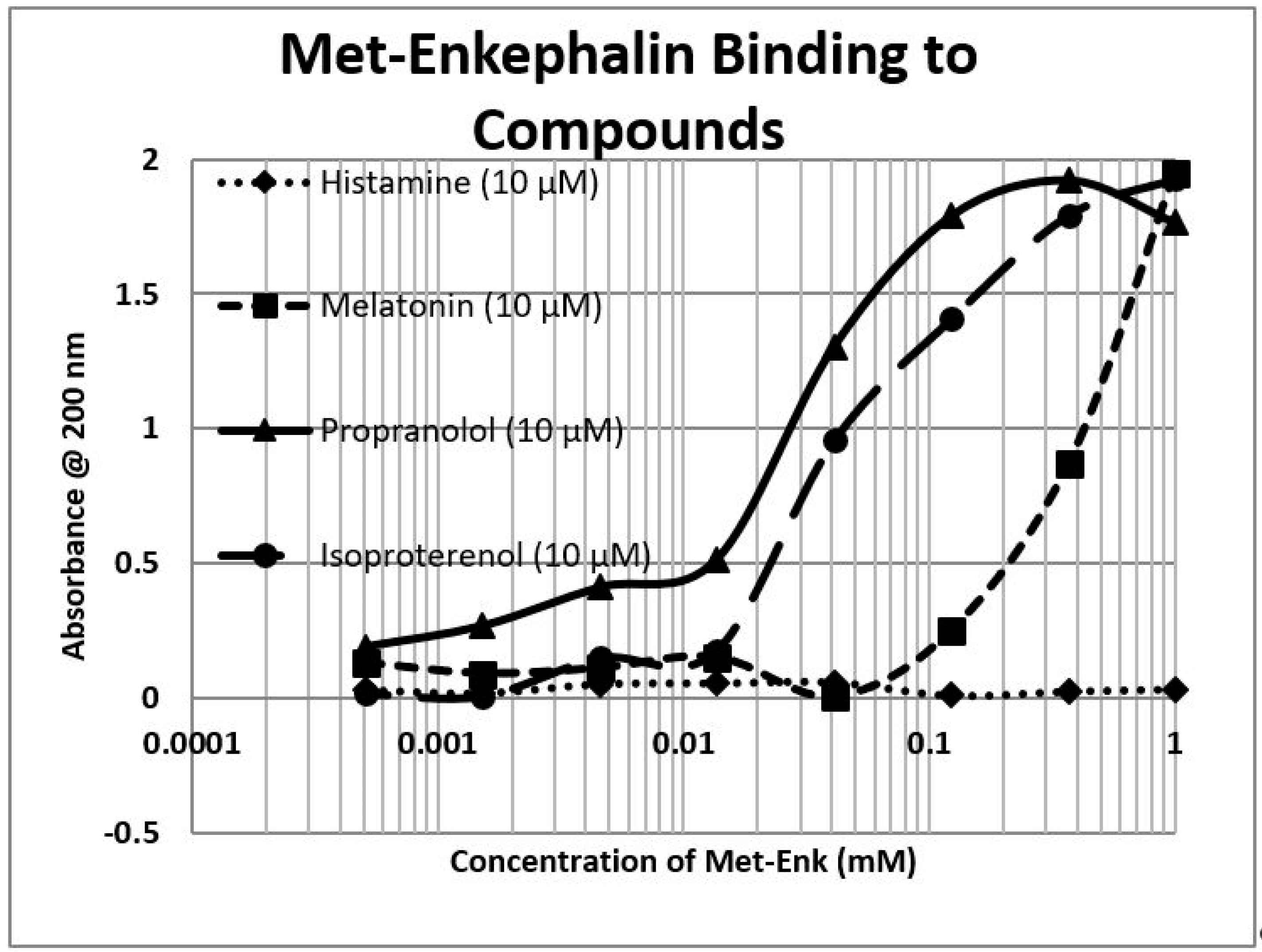
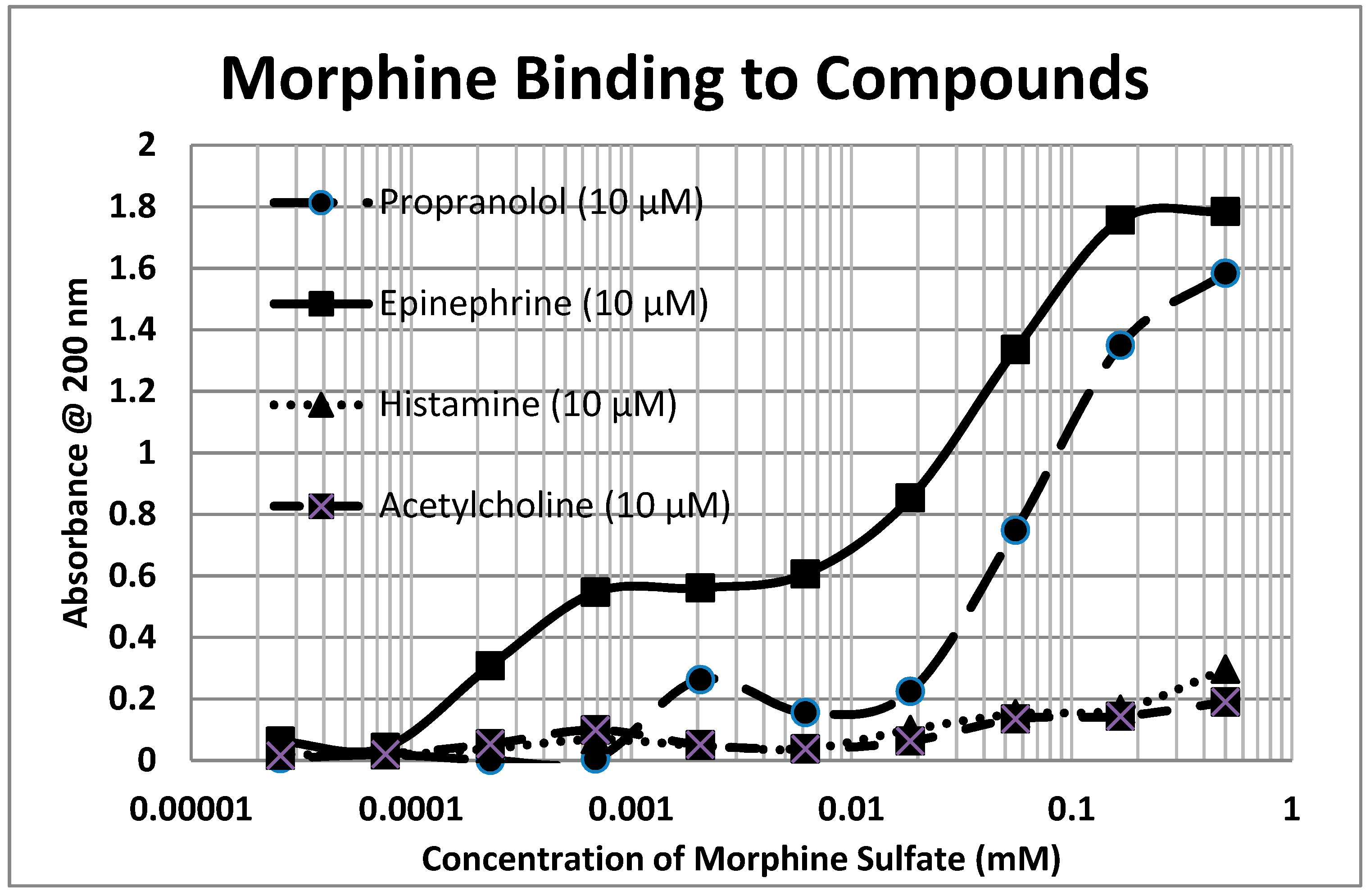
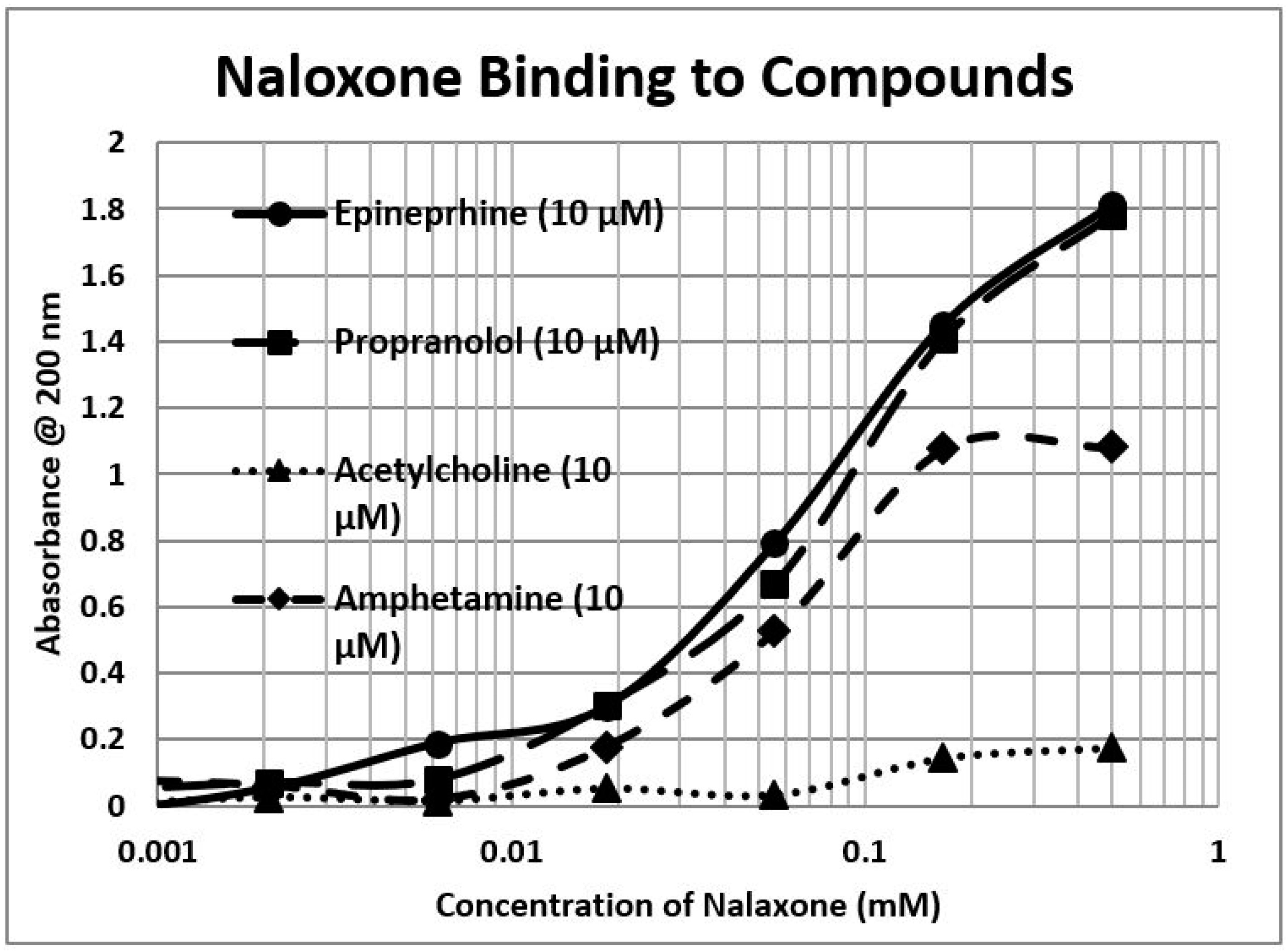
2.1. Opioid Compounds Bind to Adrenergic Compounds
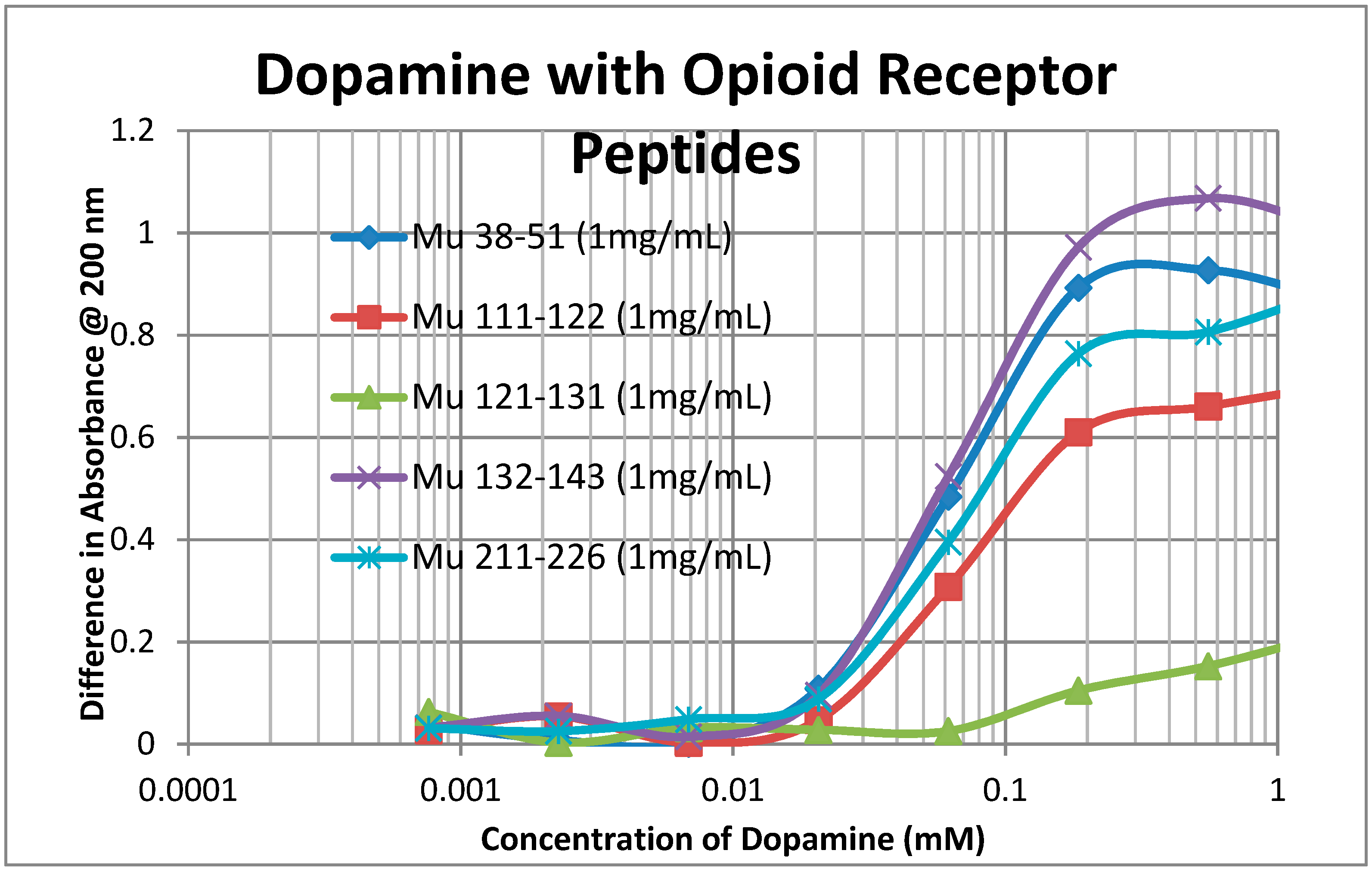
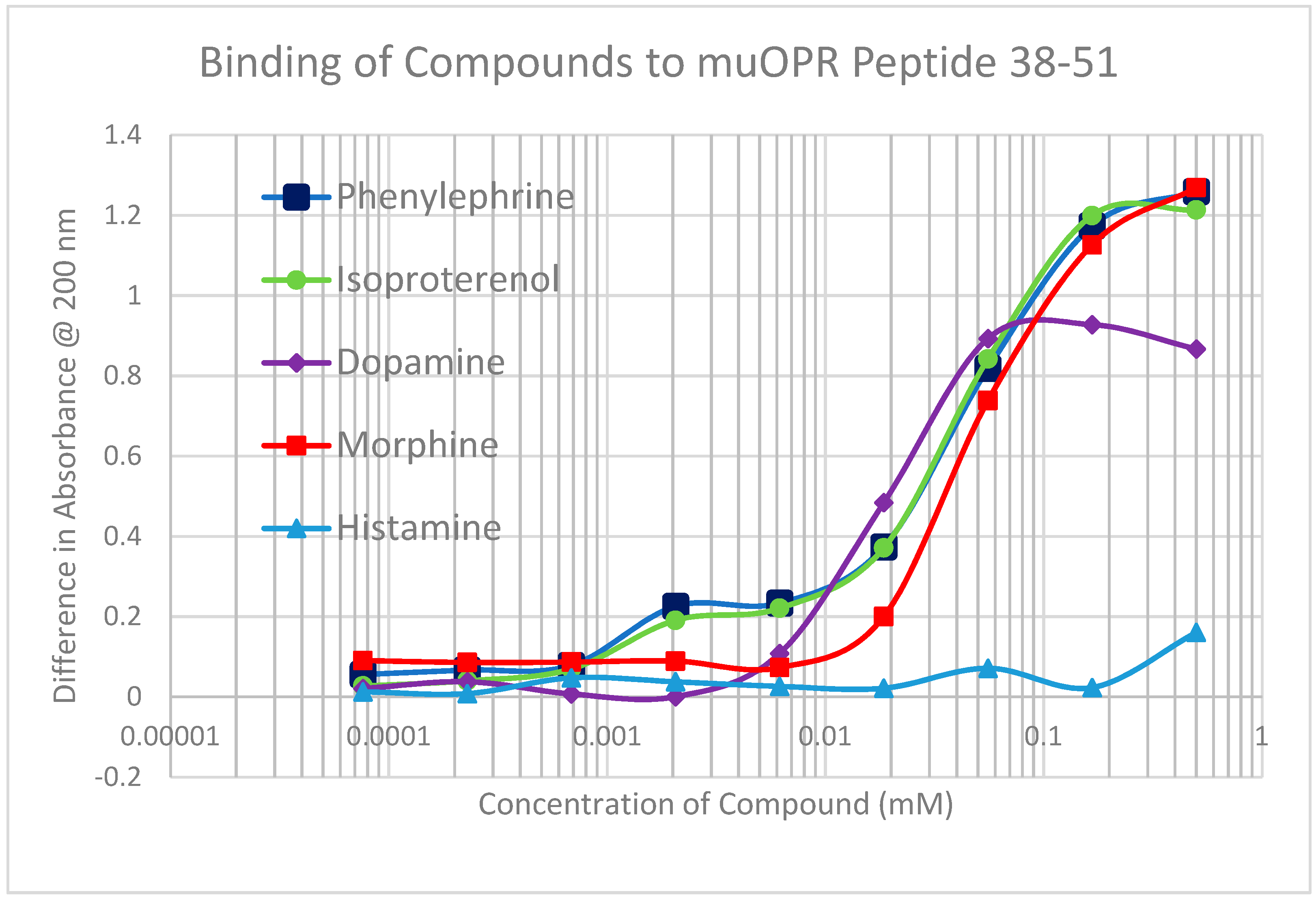
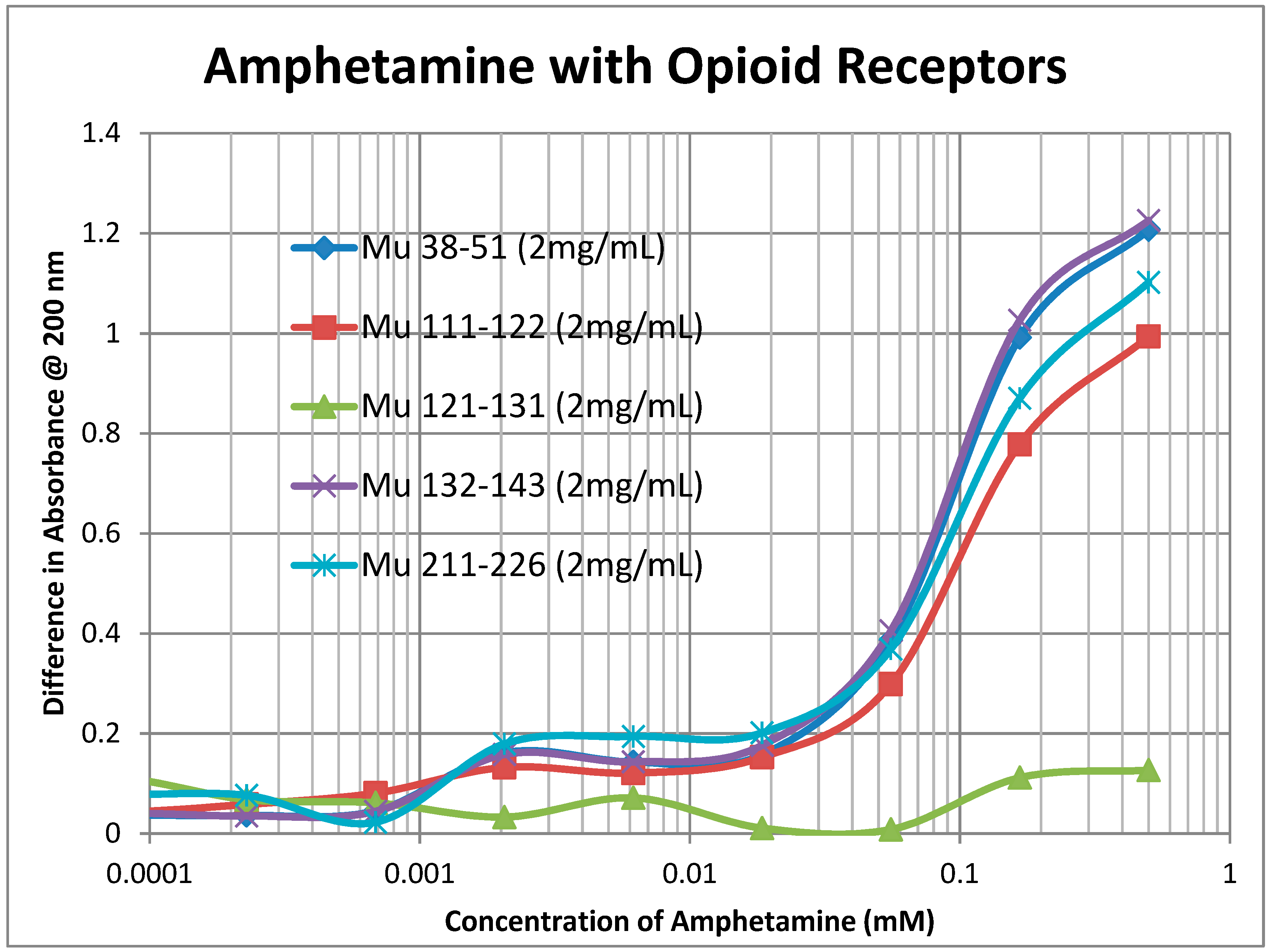
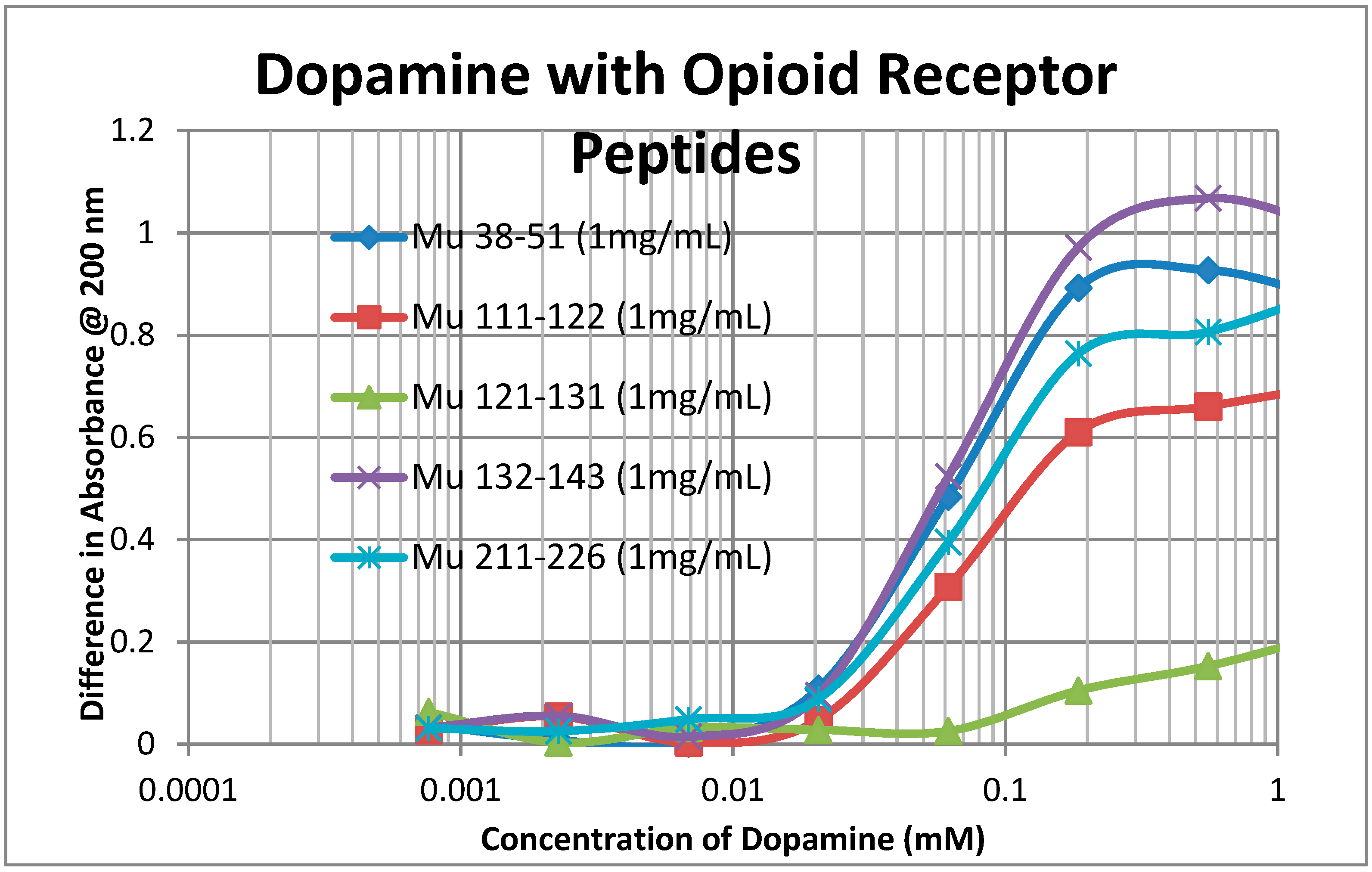
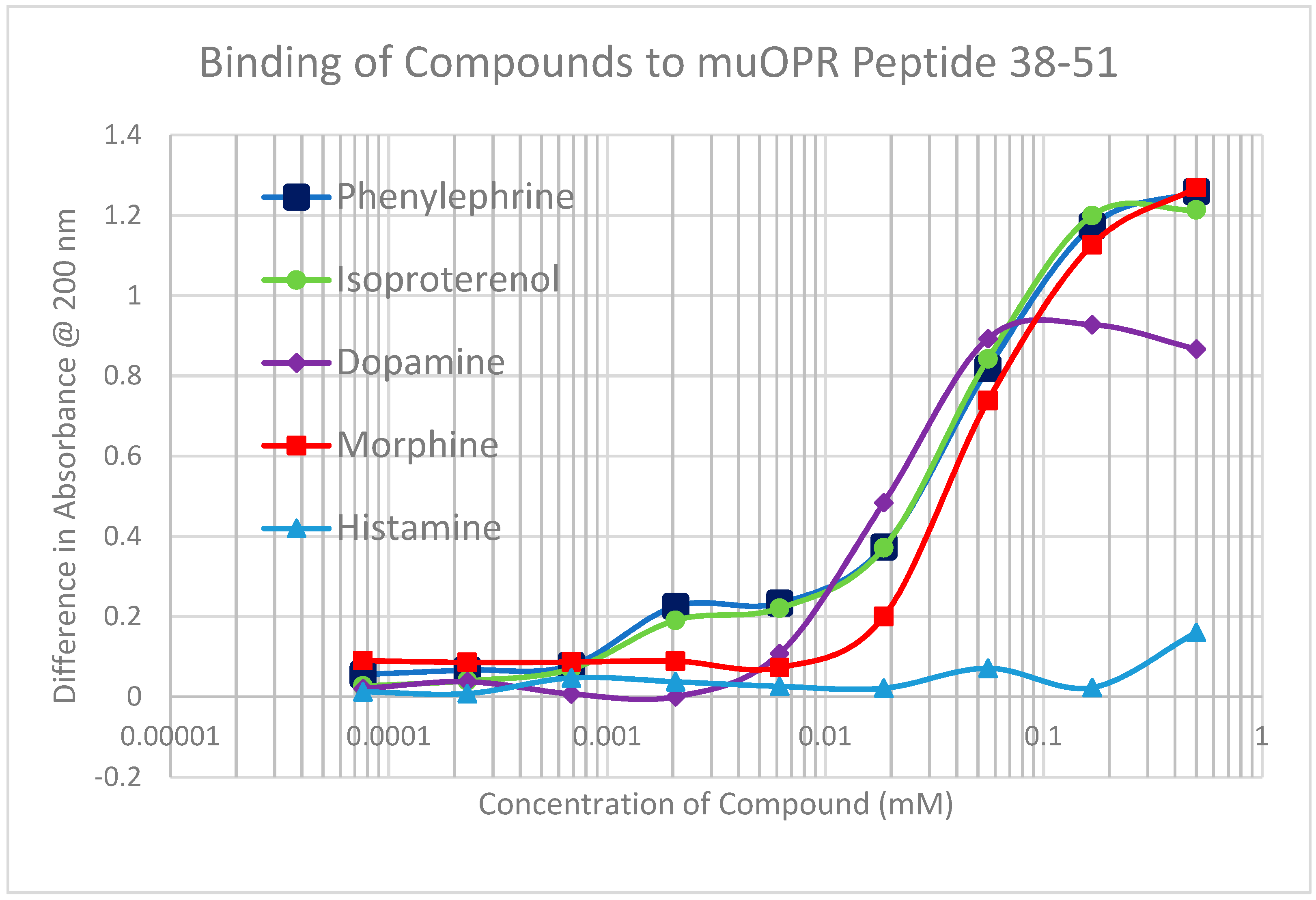
2.2. Adrenergic Compounds Bind to Opioid Receptor Peptides
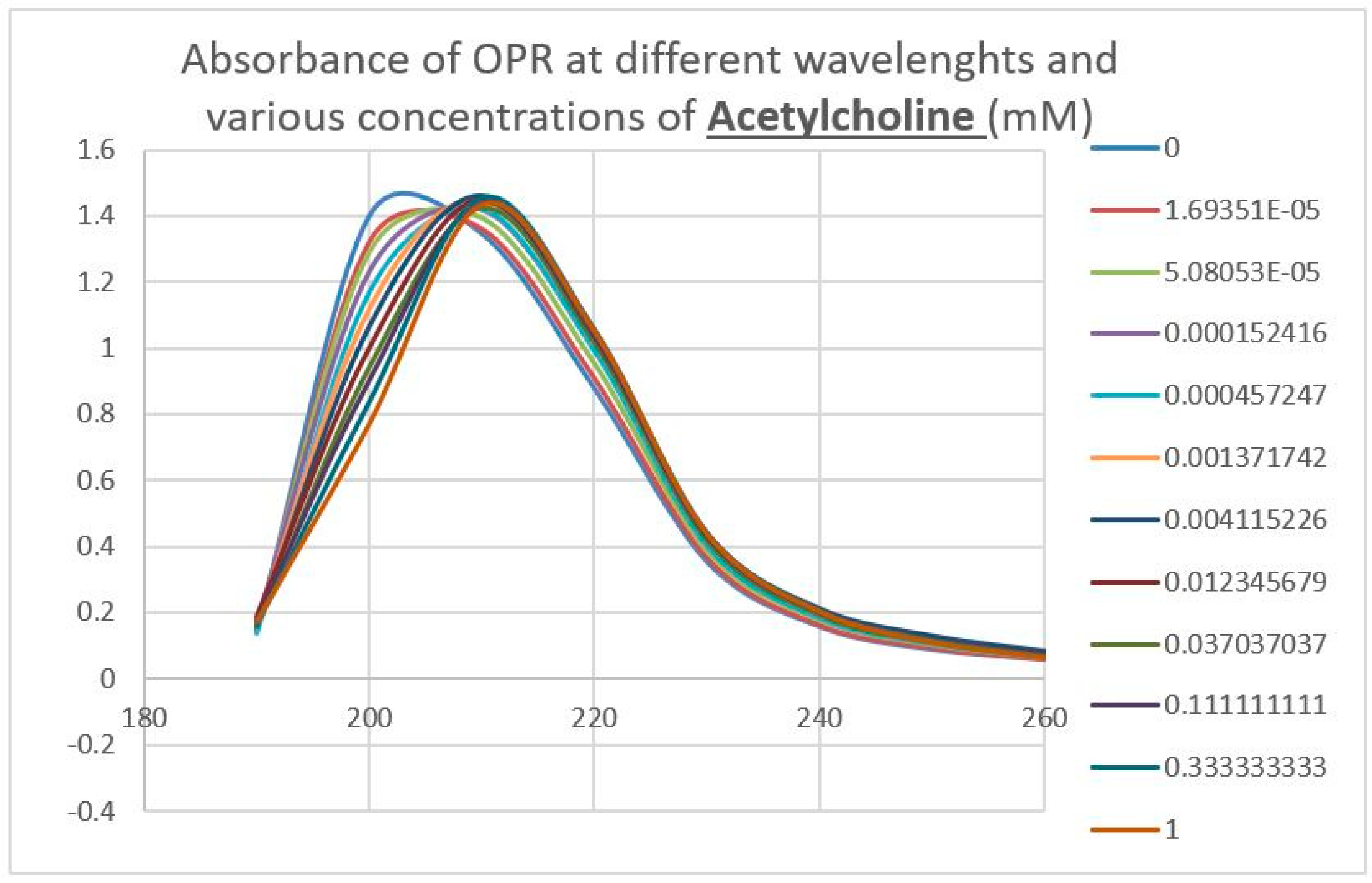
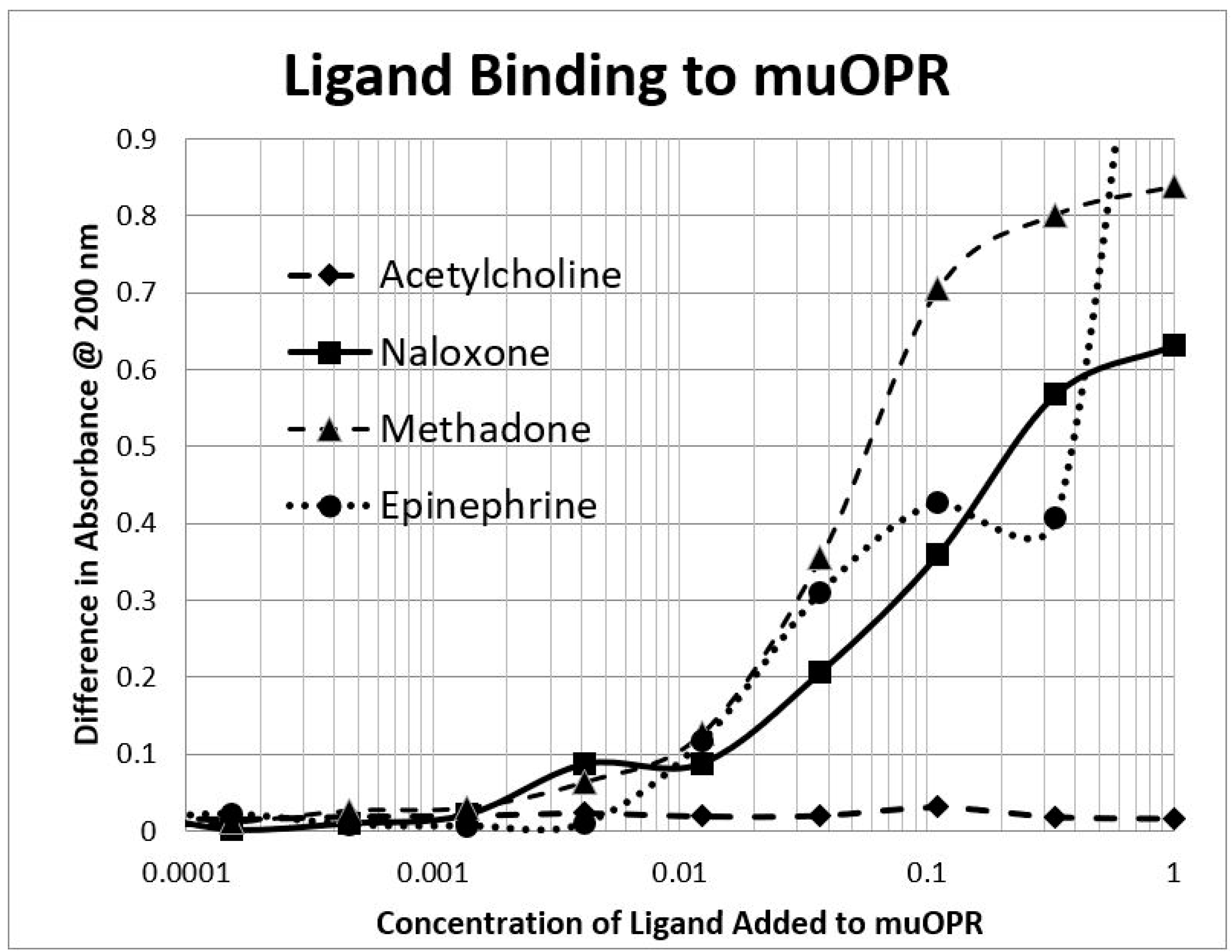
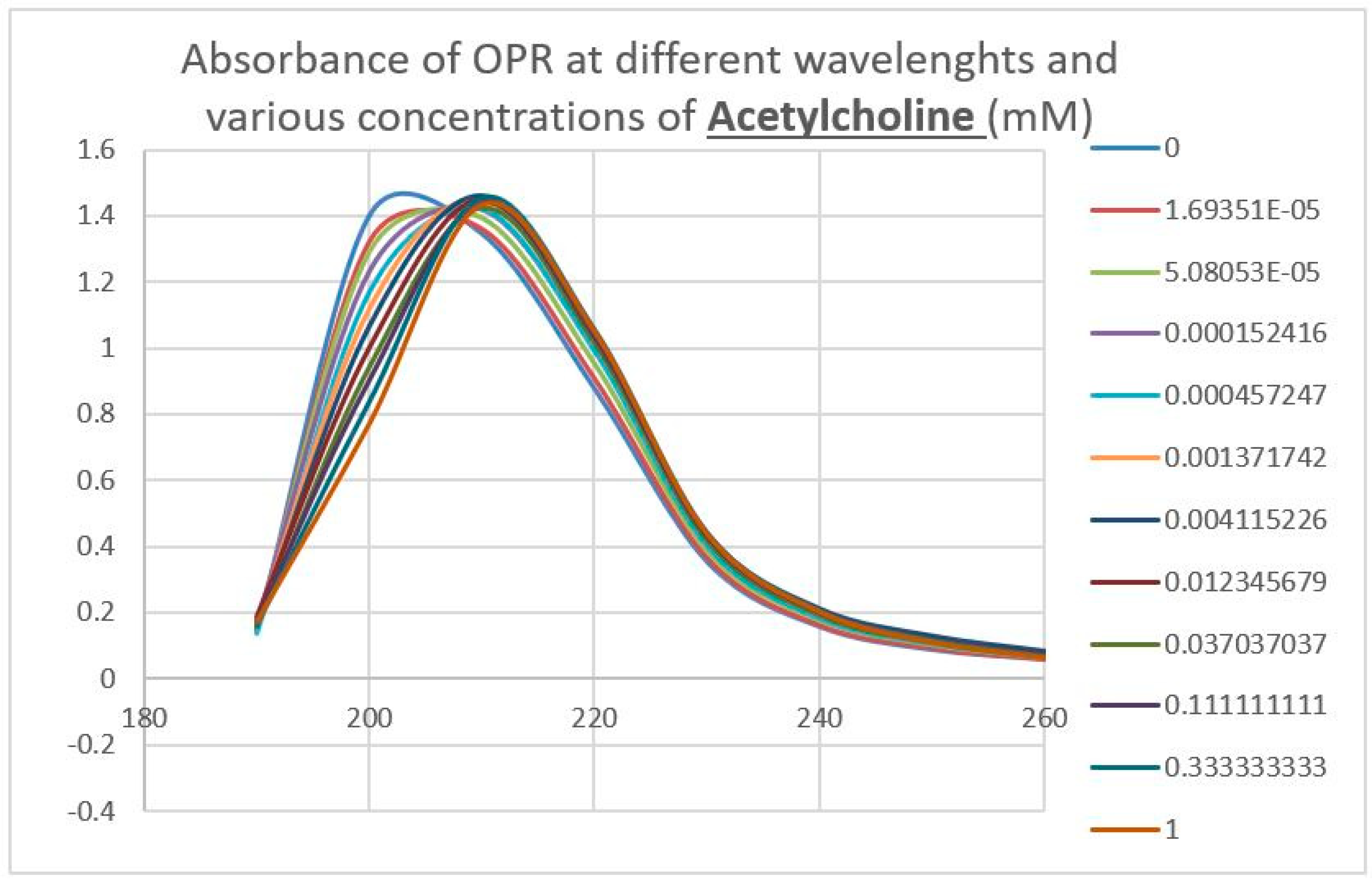
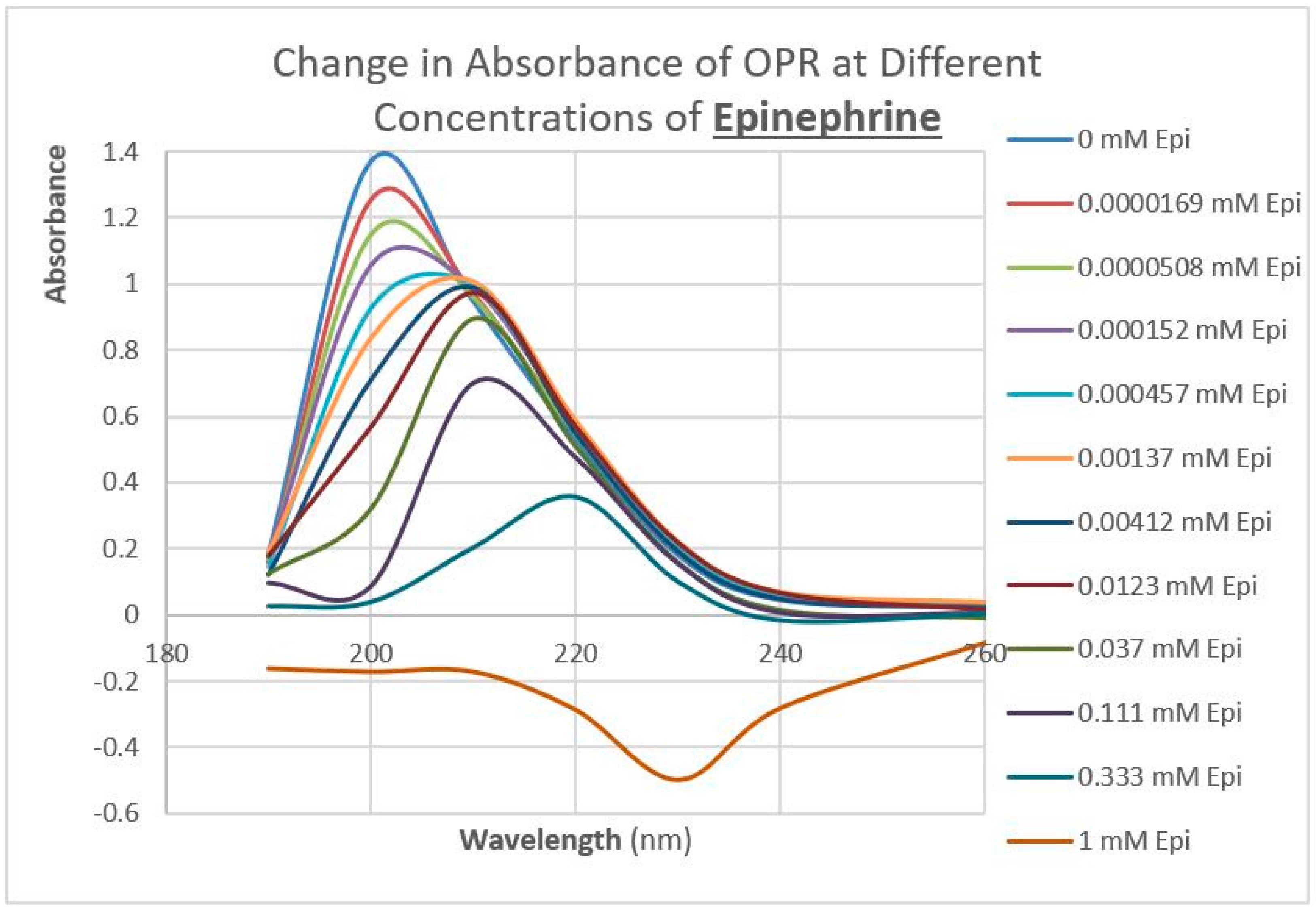
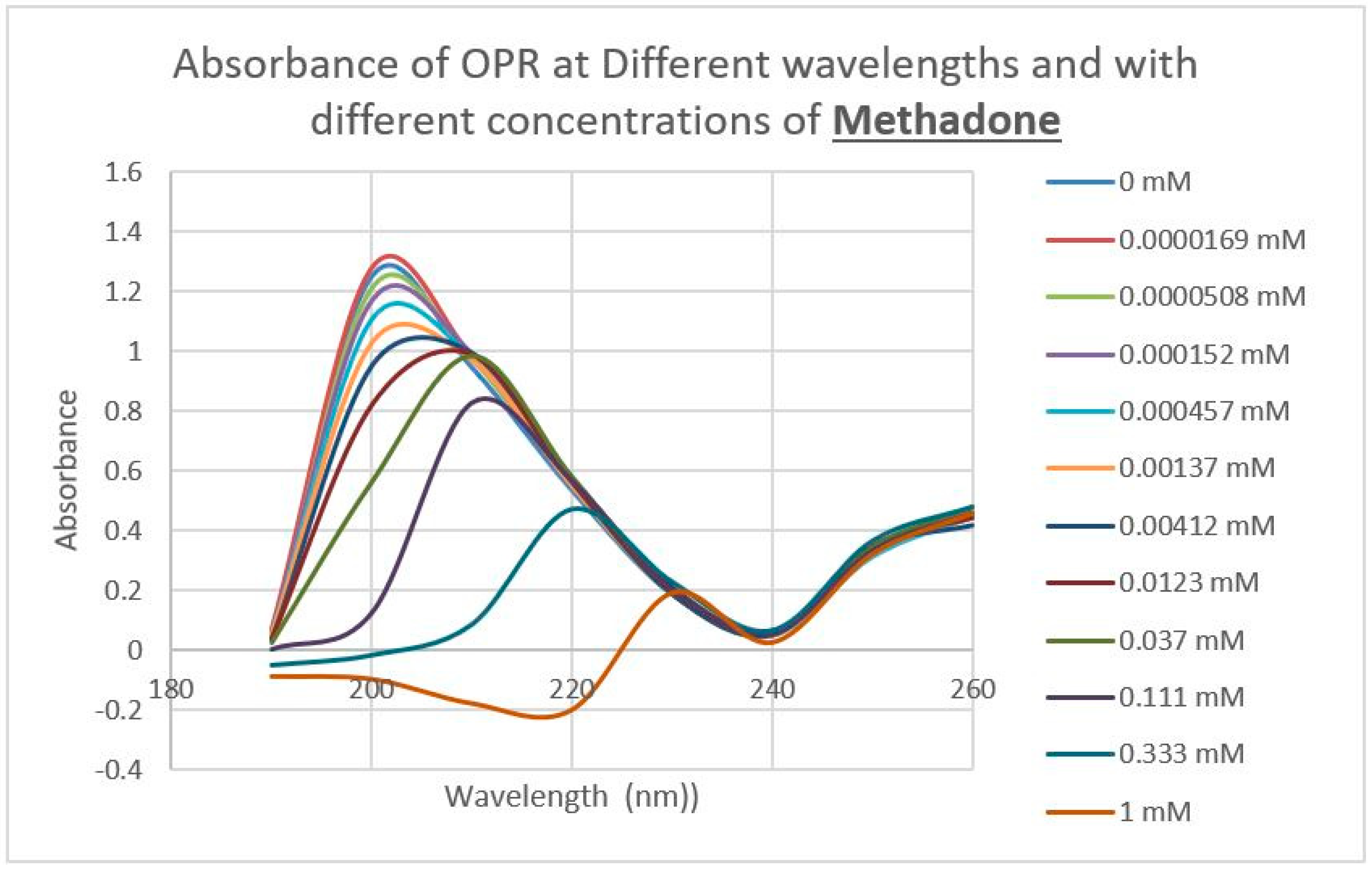
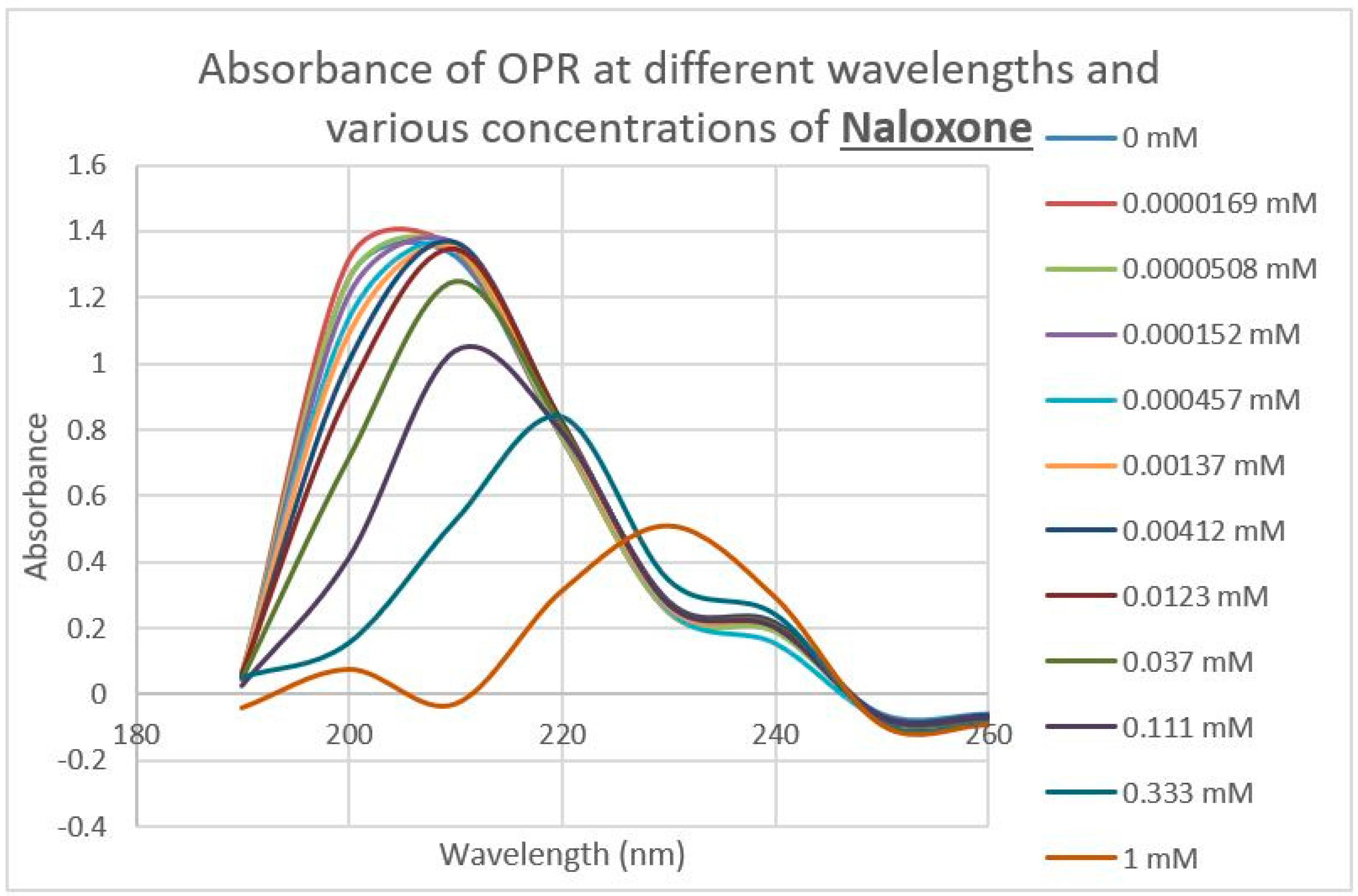
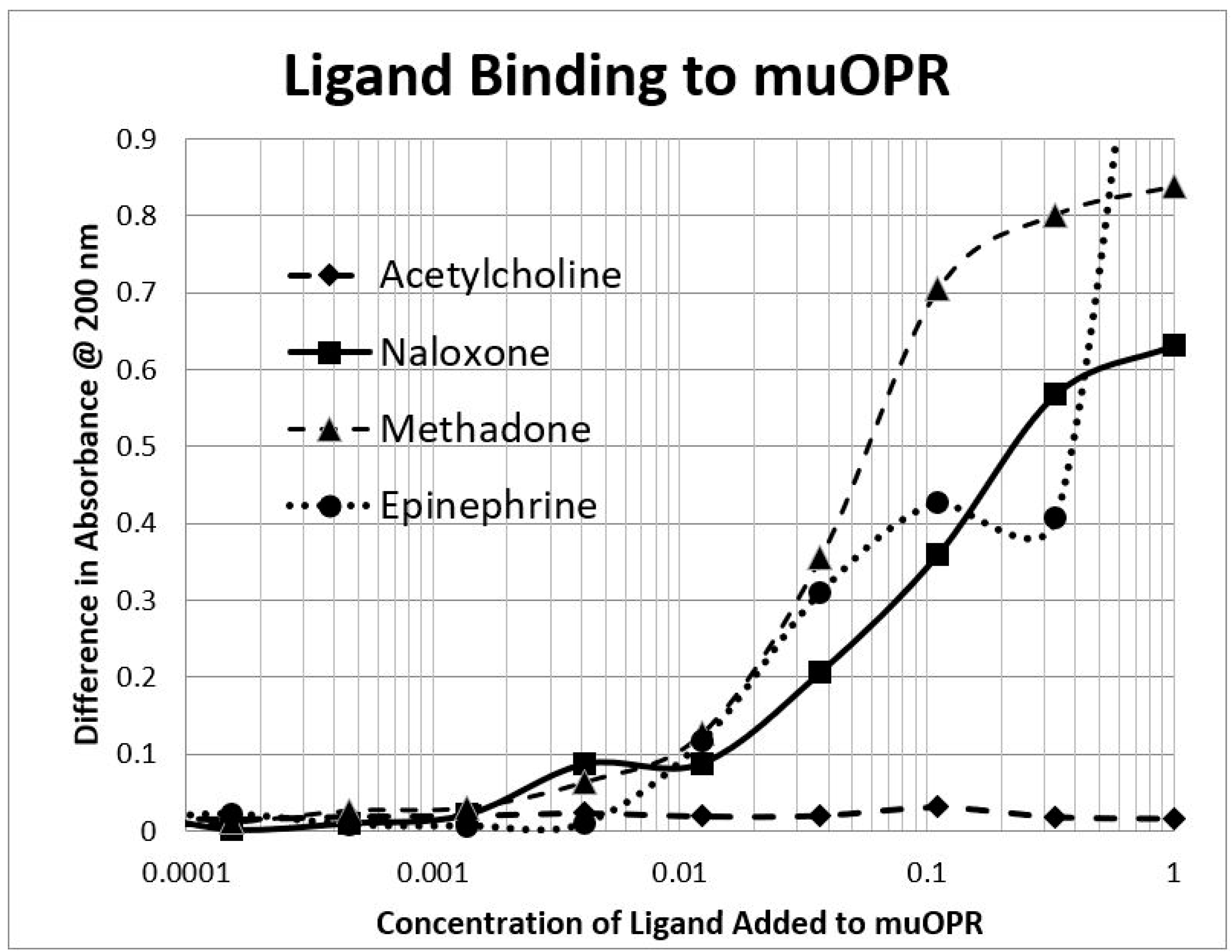
2.3. Epinephrine Binds to Intact Mu Opioid Receptor
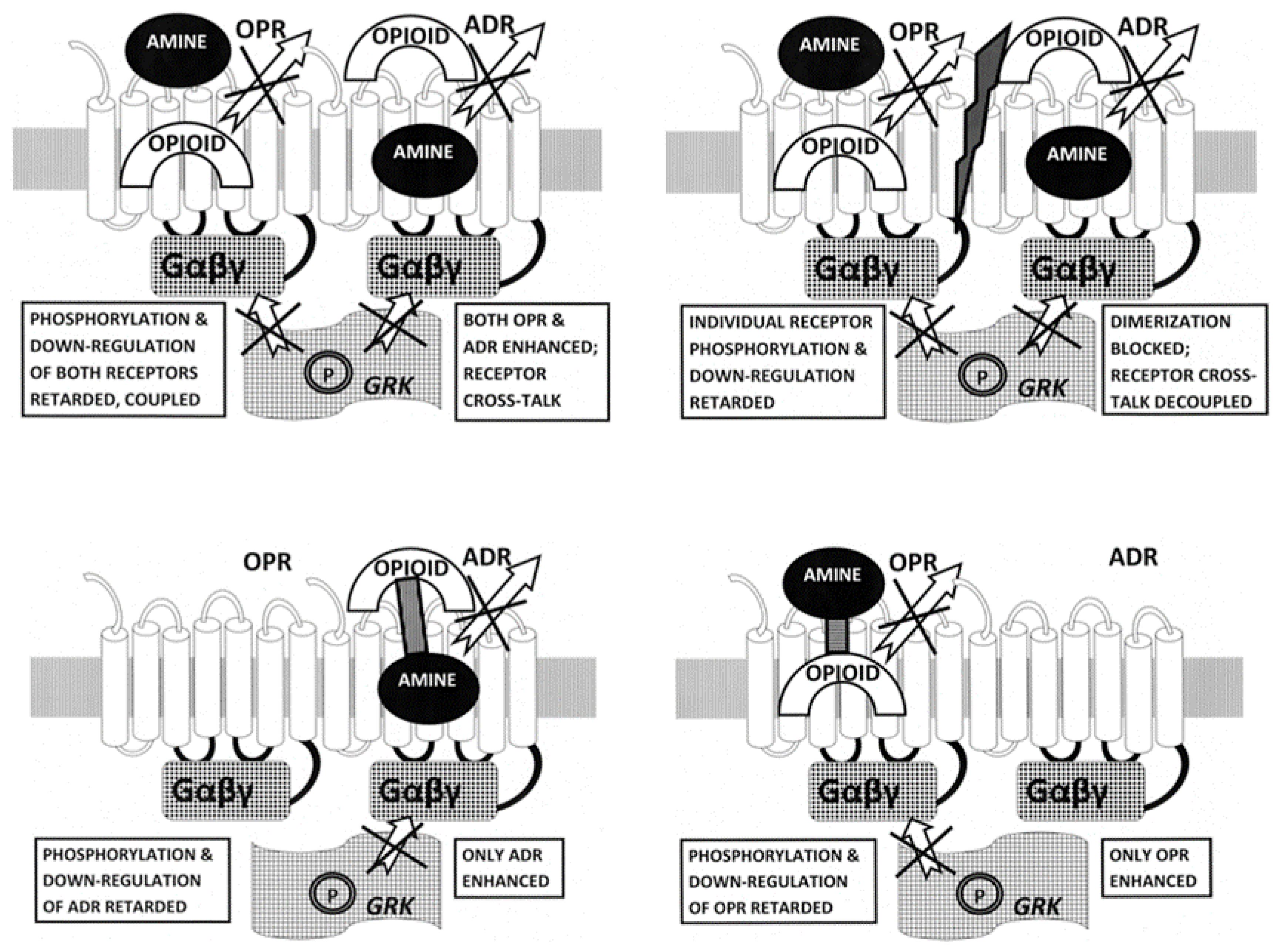
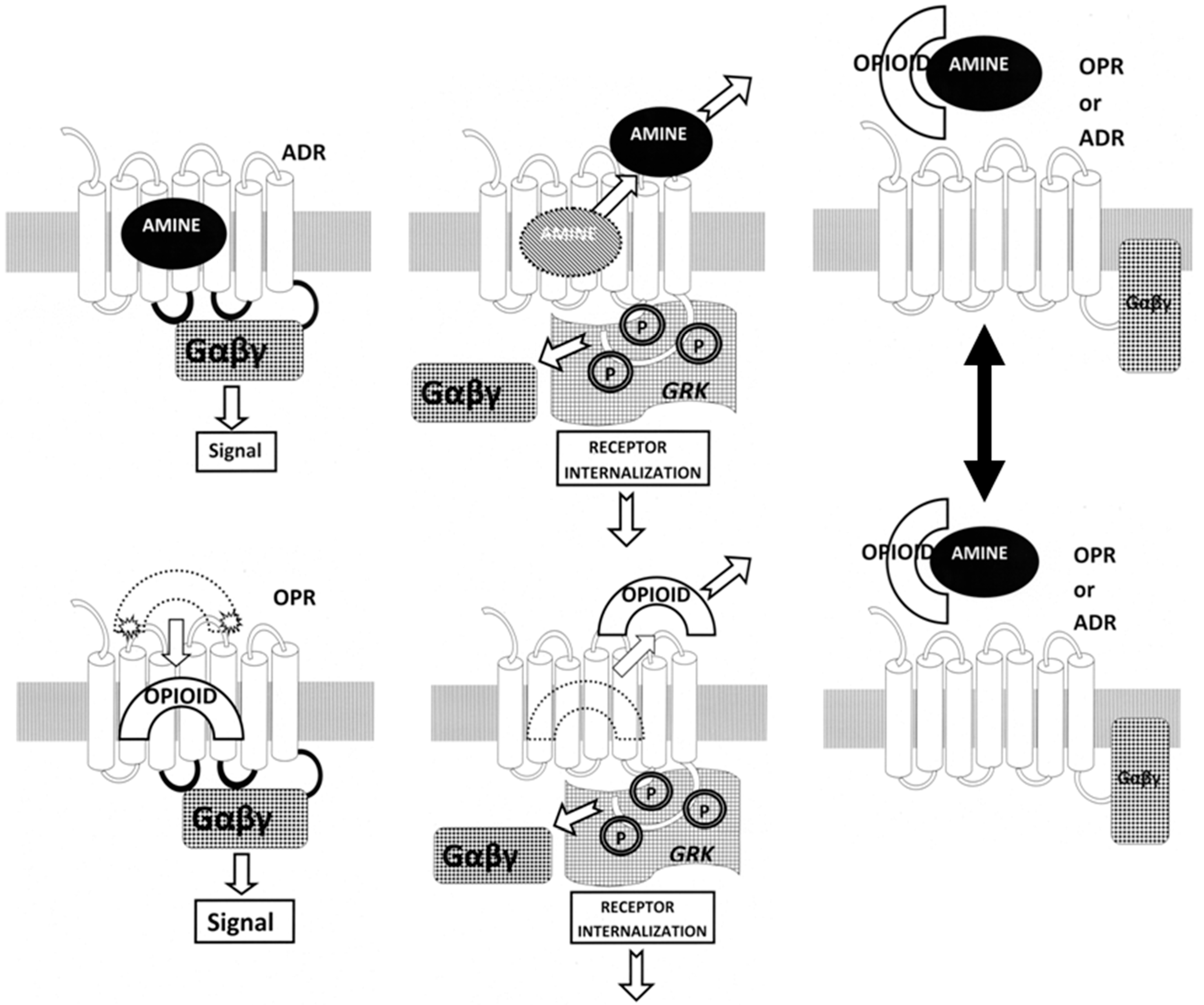
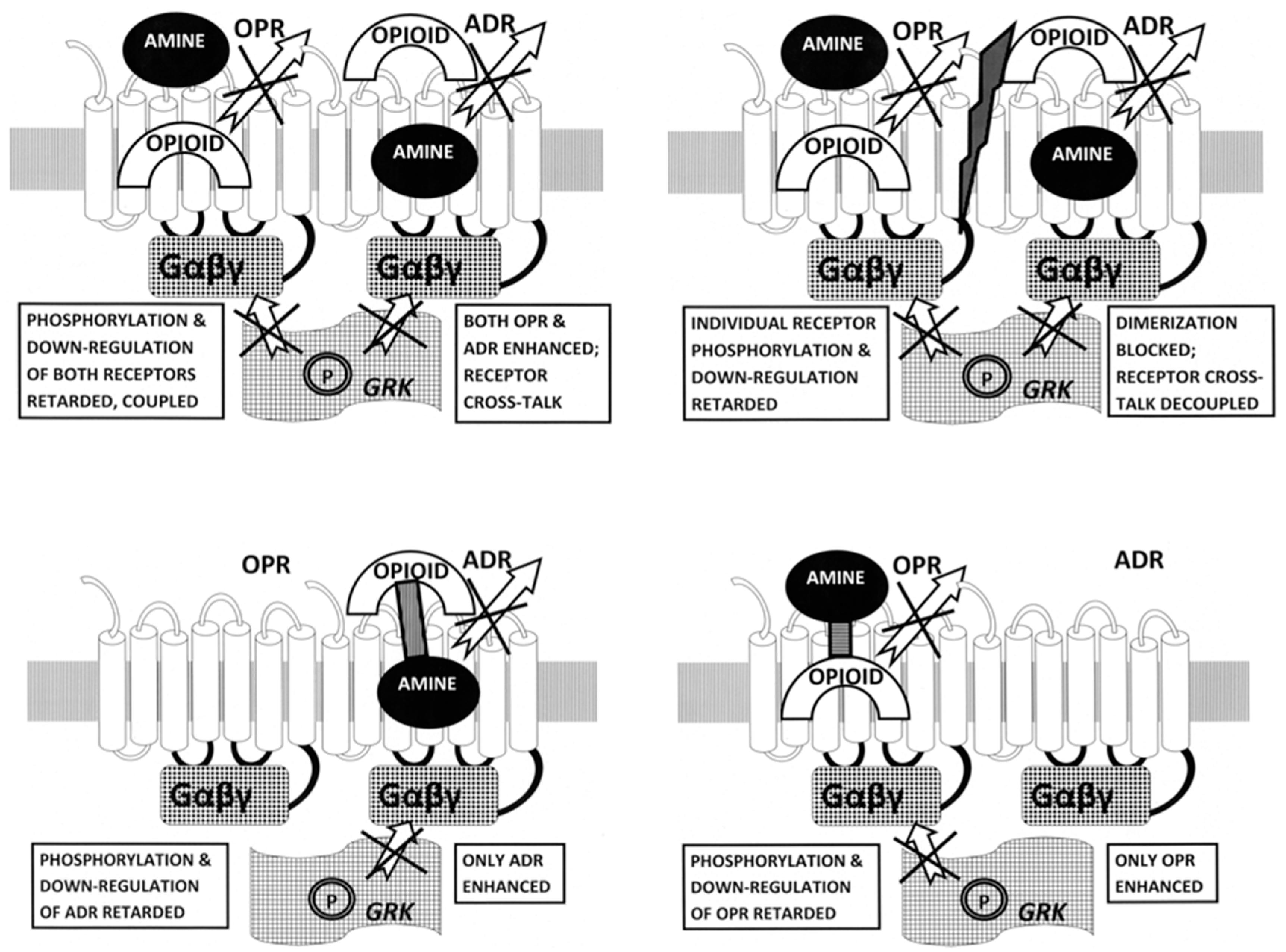
3. Discussion
3.1. Summary of Observations
3.2. Ligand Complementarity and Drug Development
3.3. Proofs of Concept for Complementary Ligand Approach to Enhancer Drug Development
4. Methods
4.1. Opioid–Adrenergic Compound Binding Test Methods
4.2. Opioid Peptide Binding Test Methods
4.3. Human Mu Opioid Receptor (muOPR) Expression and Purification
4.4. Binding of Epinephrine and Opioids to muOPR Monitored by Ultraviolet Spectroscopy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schultzberg, M.; Hökfelt, T.; Lundberg, J.M.; Terenius, L.; Elfvin, L.G.; Elde, R. Enkephalin-like immunoreactivity in nerve terminals in sympathetic ganglia and adrenal medulla and in adrenal medullary gland cells. Acta Physiol. Scand. 1978, 103, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Charnay, Y.; Leger, L.; Rossier, J.; Jouvet, M.; Dubois, P.M. Evidence for synenkephalin-like immunoreactivity in pontobulbar monoaminergic neurons of the cat. Brain Res. 1985, 335, 160–164. [Google Scholar] [CrossRef]
- Zhuo, H.; Fung, S.J.; Reddy, V.K.; Barnes, C.D. Immunohistochemical evidence for coexistence of methionine-enkephalin and tyrosine hydroxylase in neurons of the locus coeruleus complex projecting to the spinal cord of the cat. J. Chem. Neuroanat. 1992, 5, 1–4. [Google Scholar] [CrossRef]
- Livett, B.G.; Marley, P.D.; Wan, D.C.; Zhou, X.F. Peptide regulation of adrenal medullary function. J. Neural Transm. Suppl. 1990, 29, 77–89. [Google Scholar] [PubMed]
- Stachowiak, M.K.; Goc, A.; Hong, J.S.; Poisner, A.; Jiang, H.K.; Stachowiak, E.K. Regulation of tyrosine hydroxylase gene expression in depolarized non-transformed bovine adrenal medullary cells: Second messenger systems and promoter mechanisms. Brain Res. Mol. Brain Res. 1994, 22, 309–319. [Google Scholar] [CrossRef]
- Glass, M.J.; Pickel, V.M. α2A-adrenergic receptors are present in μ-opioid receptor containing neurons in rat medial nucleus tractus solitarius. Synapse 2002, 43, 208–218. [Google Scholar] [CrossRef]
- Riedl, M.S.; Schnell, S.A.; Overland, A.C.; Chabot-Doré, A.J.; Taylor, A.M.; Ribeiro-da-Silva, A.; Elde, R.P.; Wilcox, G.L.; Stone, L.S. Coexpression of α 2A-adrenergic and δ-opioid receptors in substance P-containing terminals in rat dorsal horn. J. Comp. Neurol. 2009, 513, 385–398. [Google Scholar] [CrossRef]
- Carr, D.J.; Gebhardt, B.M.; Paul, D. αAdrenergic and μ-2 opioid receptors are involved in morphine-induced suppression of splenocyte natural killer activity. J. Pharmacol. Exp. Ther. 1993, 264, 1179–1186. [Google Scholar]
- Jordan, B.A.; Trapaidze, N.; Gomes, I.; Nivarthi, R.; Devi, L.A. Oligomerization of opioid receptors with β 2-adrenergic receptors: A role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar]
- Jordan, B.A.; Gomes, I.; Rios, C.; Filipovska, J.; Devi, L.A. Functional interactions between mu opioid and α 2A-adrenergic receptors. Mol. Pharmacol. 2003, 64, 1317–1324. [Google Scholar] [CrossRef]
- Rozenfeld, R.; Devi, L.A. Exploring a role for heteromerization in GPCR signaling specificity. Biochem. J. 2011, 433, 11–18. [Google Scholar] [CrossRef]
- Vilardaga, J.-P.; Nikolaev, O.V.; Lorentz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Direct inhibition of G protein signaling by cross-conformational switches between a2A-adrenergic and µ-opioid receptors. Nat. Chem. Biol. 2008, 4, 126–131. [Google Scholar] [CrossRef]
- Vilardaga, J.-P.; Agnati, L.F.; Fuxe, K.; Ciruela, F. G-protein-coupled receptor heteromer dynamics. J. Cell Sci. 2010, 123, 4215–4220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Limbird, L.E. Hetero-oligomers of α2A-adrenergic and μ-opioid receptors do not lead to transactivation of G-proteins or altered endocytosis profiles. Biochem. Soc. Trans. 2004, 32, 856–860. [Google Scholar] [CrossRef]
- Fujita, W.; Gomes, I.; Devi, L.A. Revolution in GPCR signalling: Opioid receptor heteromers as novel therapeutic targets: IUPHAR review 10. Br. J. Pharmacol. 2014, 171, 4155–4176. [Google Scholar] [CrossRef]
- Chabot-Doré, A.J.; Millecamps, M.; Naso, L.; Devost, D.; Trieu, P.; Piltonen, M.; Diatchenko, L.; Fairbanks, C.A.; Wilcox, G.L.; Hébert, T.E.; et al. Dual allosteric modulation of opioid antinociceptive potency by α2A-adrenoceptors. Neuropharmacology 2015, 99, 285–300. [Google Scholar] [CrossRef]
- Drouin, C.; Darracq, L.; Trovero, F.; Blanc, G.; Glowinski, J.; Cotecchia, S.; Tassin, J.P. α1b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J. Neurosci. 2002, 22, 2873–2884. [Google Scholar] [CrossRef]
- Auclair, A.; Drouin, C.; Cotecchia, S.; Glowinski, J.; Tassin, J.P. 5-HT2A and α1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur. J. Neurosci. 2004, 20, 3073–3084. [Google Scholar] [CrossRef]
- Kanigel, R. Apprentice to Genius; Macmillan: New York, NY, USA, 1986; pp. 171–201. [Google Scholar]
- Munro, T.A.; Huang, X.-P.; Inglese, C.; Perrone, M.G.; Van’t Veer, A.; Carroll, F.I.; Béguin, C.; Carlezon, W.A., Jr.; Colabufo, N.A.; Cohen, B.M.; et al. Selective κ Opioid Antagonists nor-BNI, GNTI and JDTic Have Low Affinities for Non-Opioid Receptors and Transporters. PLoS ONE 2013, 8, e70701. [Google Scholar] [CrossRef]
- Lengyel, I.; Toth, F.; Biyashev, D.; Szatmari, I.; Monory, K.; Tomboly, C.; Toth, G.; Benyhe, S.; Borsodi, A. A novel non-opioid binding site for endomorphin-1. J. Physiol. Pharmacol. 2016, 67, 605–616. [Google Scholar]
- He, J.-R.; Molnar, J.; Barraclough, C.A. Morphine amplifies norepinephrine (NE)-induced LH release but blocks NE-stimulated increases in LHRH mRNA levels: Comparison of responses obtained in ovariectomized, estrogen-treated normal and androgen-sterilized rats. Mol. Brain Res. 1993, 20, 71–78. [Google Scholar] [CrossRef]
- Kindman, L.A.; Kates, R.E.; Ginsburg, R. Opioids potentiate contractile response of rabbit myocardium to the β adrenergic agonist isoproterenol. J. Cardiovasc. Pharmacol. 1991, 17, 61–67. [Google Scholar] [CrossRef]
- Lechner, R.B.; Gurll, N.J.; Reynolds, D.G. Naloxone potentiates the cardiovascular effects of catecholamines in canine hemorrhagic shock. Circ. Shock 1985, 16, 347–361. [Google Scholar]
- He, J.-R.; Barraclough, C.A. Morphine but not naloxone enhances luteinizing hormone-releasing hormone neuronal responsiveness to norepinephrine. J. Neuroendocrinol. 1992, 4, 92–99. [Google Scholar] [CrossRef]
- Allgood, S.C.; Gurll, N.J.; Reynolds, D.G. Naloxone requires circulating catecholamines to attenuate the cardiovascular suppression of endotoxic shock. J. Surg. Res. 1988, 44, 73–81. [Google Scholar] [CrossRef]
- Caffrey, J.L.; Hathorne, L.F.; Carter, G.C.; Sinclair, R.J. Naloxone potentiates contractile responses to epinephrine in isolated canine arteries. Circ. Shock 1990, 31, 317–332. [Google Scholar]
- Caffrey, J.L.; Stoll, S.T.; Sinclair, R.J.; Barron, B.A. (+) naloxone enhances vascular contractile responses to added epinephrine. Prog. Clin. Biol. Res. 1990, 328, 375–378. [Google Scholar]
- Lechner, R.B. Naloxone potentiates inotropic but not chronotropic effects of isoproterenol in vitro. Circ. Shock 1993, 39, 226–230. [Google Scholar] [CrossRef]
- Gu, H.; Gaugl, J.F.; Barron, B.A.; Caffrey, J.L. Naloxone enhances cardiac contractile responses to epinephrine without altering epinephrine uptake from plasma. Circ. Shock 1990, 32, 257–271. [Google Scholar]
- McCubbin, J.A.; Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; Feinglos, M.N. Naltrexone potentiates glycemic responses during stress and epinephrine challenge in genetically obese mice. Psychosom. Med. 1989, 51, 441–448. [Google Scholar] [CrossRef]
- Parra, L.; Pérez-Vizcaíno, F.; Alsasua, A.; Martín, M.I.; Tamargo, J. Mu- and delta-opioid receptor-mediated contractile effects on rat aortic vascular smooth muscle. Eur J Pharmacol. 1995, 277(1), 99–105. [Google Scholar] [CrossRef]
- Lee, C.H.; Berkowitz, B.A. Stereoselective and calcium-dependent contractile effects of narcotic antagonist analgesics in the vascular smooth muscle of the rat. J. Pharmacol. Exp. Ther. 1976, 198, 347–356. [Google Scholar]
- Lee, C.H.; Berkowitz, B.A. Calcium antagonist activity of methadone, L-acetylmethadol and L-pentazocine in the rat aortic strip. J. Pharmacol. Exp. Ther. 1977, 202, 646–653. [Google Scholar]
- Deyo, S.N.; Swift, R.M.; Miller, R.J. Morphine and endorphins modulate dopamine turnover in rat median eminence. Proc. Natl. Acad. Sci. USA 1979, 76, 3006–3009. [Google Scholar] [CrossRef]
- Deyo, S.N.; Swift, R.M.; Miller, R.J.; Fang, V.S. Development of tolerance to the prolactin-releasing action of morphine and its modulation by hypothalamic dopamine. Endocrinology 1980, 106, 1469–1474. [Google Scholar] [CrossRef]
- Tagaya, E.; Tamaoki, J.; Chiyotani, A.; Konno, K. Stimulation of opioid mu-receptors potentiates β adrenoceptor-mediated relaxation of canine airway smooth muscle. J. Pharmacol. Exp. Ther. 1995, 275, 1288–1292. [Google Scholar]
- Roerig, S.C.; Lei, S.; Kitto, K.; Hylden, J.K.; Wilcox, G.L. Spinal interactions between opioid and noradrenergic agonists in mice: Multiplicativity involves delta and alpha-2 receptors. J. Pharmacol. Exp. Ther. 1992, 262, 365–374. [Google Scholar]
- Root-Bernstein, R.S.; Dillon, P.F. Fostering adventure research. A case study of the discovery that ascorbic acid enhances adrenergic drug activity. Drug Dev. Res. 2002, 57, 58–74. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Dillon, P.F. A common molecular motif characterizes extracellular allosteric enhancers of GPCR aminergic receptors and suggests their mechanism of action. Curr. Med. Chem. 2014, 21, 3673–3686. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Fewins, J.; Dillon, P.F. Tartaric Acid Enhances Adrenergic Receptor Activity: Test of a General Theory of Extracellular Aminergic GPCR Enhancer Discovery. Curr. Drug Discov. Technol. 2014, 11, 293–307. [Google Scholar] [CrossRef]
- Abel, S.; Harris, S.C. Morphine-benzedrine analgesia in obstetrics. Fed Proc. 1947, 6, 67. [Google Scholar]
- Milosevic, M.P. Effect of adrenaline on the analgesic response of mice to morphine and related drugs. Arch. Int. Pharmacodyn. Ther. 1955, 104, 50–56. [Google Scholar]
- Goyagi, T.; Nishikawa, T. The addition of epinephrine enhances postoperative analgesia by intrathecal morphine. Anesth. Analg. 1995, 81, 508–513. [Google Scholar]
- Goyagi, T.; Nishikawa, T. Oral clonidine premedication enhances the quality of postoperative analgesia by intrathecal morphine. Anesth Analg. 1996, 82, 1192–1196. [Google Scholar]
- Sasson, S.; Unterwald, E.M.; Kornetsky, C. Potentiation of morphine analgesia by d-amphetamine. Psychopharmacology 1986, 90, 163–165. [Google Scholar] [CrossRef]
- Izenwasser, S.; Kornetsky, C. Potentiation of morphine analgesia by d-amphetamine is mediated by norepinephrine and not dopamine. Pain 1988, 33, 363–368. [Google Scholar] [CrossRef]
- Huang, K.S.; Tseng, C.H.; Cheung, K.S.; Hui, Y.L.; Tan, P.P. Influence of epinephrine as an adjuvant to epidural morphine for postoperative analgesia. Ma Zui Xue Za Zhi 1993, 31, 245–248. [Google Scholar]
- Sierralta, F.; Naquira, D.; Pinardi, G.; Miranda, H.F. α-Adrenoceptor and opioid receptor modulation of clonidine-induced antinociception. Br. J. Pharmacol. 1996, 119, 551–554. [Google Scholar] [CrossRef]
- Wu, Y.W.; Seah, Y.S.; Chung, K.T.; Liu, M.D. Postoperative pain relief in primigravida caesarean section patients—Combination of intrathecal morphine and epinephrine. Acta Anaesthesiol. Sin. 1999, 37, 111–114. [Google Scholar]
- Gulati, A.; Bhalla, S.; Matwyshyn, G.; Zhang, Z.; Andurkar, S.V. Determination of adrenergic and imidazoline receptor involvement in augmentation of morphine and oxycodone analgesia by clonidine and BMS182874. Pharmacology 2009, 83, 45–58. [Google Scholar] [CrossRef]
- Fairbanks, C.A.; Posthumus, I.J.; Kitto, K.F.; Stone, L.S.; Wilcox, G.L. Moxonidine, a selective imidazoline/α2 adrenergic receptor agonist, synergizes with morphine and deltorphin II to inhibit substance P-induced behavior in mice. Pain 2000, 84, 13–20. [Google Scholar] [CrossRef]
- Gupta, S.; Raval, D.; Patel, M.; Patel, N.; Shah, N. Addition of epidural Clonidine enhances postoperative analgesia: A double-blind study in total knee- replacement surgeries. Anesth. Essays Res. 2010, 4, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Engelman, E.; Marsala, C. Efficacy of adding clonidine to intrathecal morphine in acute postoperative pain: Meta-analysis. Br. J. Anaesth. 2013, 110, 21–27. [Google Scholar] [CrossRef]
- Katz, D.; Hamburger, J.; Gutman, D.; Wang, R.; Lin, H.M.; Marotta, M.; Zahn, J.; Beilin, Y. The effect of adding subarachnoid epinephrine to hyperbaric bupivacaine and morphine for repeat cesarean delivery: A double-blind prospective randomized control trial. Anesth. Analg. 2017. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Turke, M.; Subhramanyam, U.K.T.; Churchill, B.; Labahn, J. Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”? Int. J. Mol. Sci. 2018, 19, 272. [Google Scholar] [CrossRef]
- Milne, B.; Sutak, M.; Cahill, C.M.; Jhamandas, K. Low doses of α 2-adrenoceptor antagonists augment spinal morphine analgesia and inhibit development of acute and chronic tolerance. Br. J. Pharmacol. 2008, 155, 1264–1278. [Google Scholar] [CrossRef]
- Satarian, L.; Javan, M.; Fathollahi, Y. Epinephrine inhibits analgesic tolerance to intrathecal administrated morphine and increases the expression of calcium-calmodulin-dependent protein kinase IIα. Neurosci. Lett. 2008, 430, 213–217. [Google Scholar] [CrossRef]
- Heimans, R.L. Catecholamines and the actions of morphine on the guinea-pig ileum. Arch. Int. Pharmacodyn. Ther. 1975, 216, 11–18. [Google Scholar]
- Ferri, S.; Reina, R.; Santagostino, A. Dopamine and the depressant action of morphine on stimulated guinea-pig ileum. Br. J. Pharmacol. 1977, 59, 25–28. [Google Scholar] [CrossRef]
- Goldstein, A.; Schulz, R. Morphine-tolerant longitudinal muscle strip from guinea-pig ileum. Br. J. Pharmacol. 1973, 48, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Sarto, G. Modulation of the effect of morphine on the isolated guinea pig intestine by noradrenaline and serotonin. Boll. Soc. Ital. Biol. Sper. 1981, 57, 394–400. [Google Scholar]
- Park, W.K.; Chang, C.H.; Chae, J.E.; Kim, M.H.; Cho, Y.L.; Ahn, D.S. Phosphodiesterase inhibition by naloxone augments the inotropic actions of beta-adrenergic stimulation. Acta Anaesthesiol. Scand. 2009, 53, 1043–1051. [Google Scholar] [CrossRef]
- Monroe, P.J.; Perschke, S.E.; Crisp, T.; Smith, D.J. Evaluation of the interactions of serotonergic and adrenergic drugs with mu, delta, and kappa opioid binding sites. Neurosci. Lett. 1991, 133, 229–232. [Google Scholar] [CrossRef]
- Dillon, P.F.; Root-Bernstein, R.; Robinson, N.E.; Abraham, W.M.; Berney, C. Receptor-mediated enhancement of β adrenergic drug activity by ascorbate in vitro and in vivo. PLoS ONE 2010, 5, e15130. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Rhinesmith, T.; Koch, A.; Dillon, P.F. Enzymatic recycling of ascorbate by glutathione-like peptides derived from aminergic G-protein coupled receptors. J. Mol. Recog. 2016. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Dillon, P.F. Molecular complementarity, I: The molecular complementarity theory of the origin and evolution of life. J. Theor. Biol. 1997, 188, 447–479. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Molecular complementarity III. Molecular complementarity as the basis for peptide hormone evolution. A bioinformatic case study of insulin, glucagon, and gastrin. J. Theor. Biol. 2002, 218, 71–84. [Google Scholar] [CrossRef]
- Hunding, A.; Lancet, D.; Kepes, F.; Minsky, A.; Norris, V.; Raine, D.; Sriram, K.; Root-Bernstein, R. Compositional complementarity and prebiotic ecosystems in the origin of life. Bioessays 2006, 28, 399–412. [Google Scholar] [CrossRef]
- Root-Bernstein, R. A modular hierarchy-based theory of the chemical origins of life based on molecular complementarity. Acc. Chem. Res. 2012, 45, 2169–2177. [Google Scholar] [CrossRef]
- Dwyer, D.S. Amino acid sequence homology between ligands and their receptors: Potential identification of binding sites. Life Sci. 1989, 45, 421–429. [Google Scholar] [CrossRef]
- Dwyer, D.S. Molecular model of interleukin 12 that highlights amino acid sequence homologies with adhesion domains and gastrointestinal peptides. J. Mol. Graphics 1996, 14, 148–157. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Peptide self-aggregation and peptide complementarity as bases for the evolution of peptide receptors: A review. J. Mol. Recog. 2005, 18, 40–49. [Google Scholar] [CrossRef]
- Root-Bernstein, R. A modular insulin-like basis for the evolution of glucose transporters (GLUT) with implications for diabetes. Evol. Bioinform. 2007, 2, 317–331. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Vonck, J. The insulin receptor binds glucose altering the mutual affinity of insulin for its receptor. Cell. Molec. Life Sci. 2009, 6, 2721–2732. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Vonck, J. Modularity in receptor evolution. Insulin- and glucagon-like peptide modules as binding sites for insulin and glucose in the insulin receptor. J. Recept. Ligand Channel Res. 2010, 3, 87–96. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Dillon, P.F. Small molecule complementarity as a source of novel pharmaceutical agents and combination therapies. Curr. Pharm. Des. 2008, 14, 55–62. [Google Scholar] [CrossRef]
- Root Bernstein, R.S. Catecholamines bind to enkephalins, morphiceptin, and morphine. Brain Res. Bull. 1987, 18, 509–532. [Google Scholar] [CrossRef]
- Dillon, P.F.; Root-Bernstein, R.S.; Sears, P.R.; Olson, L.K. Natural electrophoresis of norepinephrine and ascorbic acid. Biophys. J. 2000, 79, 370–376. [Google Scholar] [CrossRef]
- Ventura, C.; Bastagli, L.; Bernardi, P.; Caldarera, C.M.; Guarnieri, C. Opioid receptors in rat cardiac sarcolemma: Effect of phenylephrine and isoproterenol. Biochim. Biophys. Acta 1989, 987, 69–74. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Dillon, P.F.; Hollingsworth, R. A tethered ascorbate-norepinephrine compound, 4-UT, displays long-acting adrenergic activity on rabbit aortic smooth muscle. Drug Res. Dev. 2008, 69, 242–250. [Google Scholar] [CrossRef]
- Draper, R.D.; Ingraham, L.L. The affinity of flavin semiquinones for certain aromatic compounds and disulfides. Arch. Biochem. Biophys. 1970, 139, 265–268. [Google Scholar] [CrossRef]
- Yamanaka, N. Effect of dimethyl sulphoxide on charge transfer complexes between serotonin-, tryptophan and flavin mononucleotide under low temperatures. J. Vitaminol. (Kyoto) 1971, 17, 39–42. [Google Scholar] [CrossRef]
- Haggi, E.; Blasich, N.; Gutiérrez, L.; Vázquez, G.; Criado, S.; Miskoski, S.; Ferrari, G.; Paulina Montaña, M.; García, N.A. On the generation and quenching of reactive-oxygen-species by aqueous vitamin B2 and serotonin under visible-light irradiation. J. Photochem. Photobiol. B 2012, 113, 22–28. [Google Scholar] [CrossRef]
- Wilson, J.E. Studies on the electronic nature of flavin-indole and flavin-purine complexes. Biochemistry 1966, 5, 1351–1359. [Google Scholar] [CrossRef]
- Budini, R.; Marinangeli, A. Interactions between some psychodrugs and flavins. Z. Naturforsch. B 1970, 25, 505–509. [Google Scholar] [CrossRef]
- Silberstein, S.D. Migraine: Preventive treatment. Curr. Med. Res. Opin. 2001, 17, s87–s93. [Google Scholar] [CrossRef]
- Pringsheim, T.; Davenport, W.; Mackie, G.; Worthington, I.; Aubé, M.; Christie, S.N.; Gladstone, J.; Becker, W.J. Canadian Headache Society Prophylactic Guidelines Development Group. Canadian Headache Society guideline for migraine prophylaxis. Can. J. Neurol. Sci. 2012, 39, S1–S59. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kd (µM) @ 200 nm | M-Enk | Morph | Meth | NAL | Amph | Prop | Asc | Glucose | Ribo |
|---|---|---|---|---|---|---|---|---|---|
| Epinephrine HCl | 5.8/40 * | 0.3/40 * | 300 | 100 | 4.5 | 45/1000 | 90 # | >1000 | >1000 |
| Norepinephrine HCl | 5.3/35 * | 0.4 * | 70 | 80 | 80 | >1000 | 110 # | >1000 | >1000 |
| Dopamine | 30 * | 0.6 * | 200 | 90 | 90 | 100 | 100 | >1000 | >1000 |
| L-DOPA | 70 * | >1000 * | 80 | 55 | 11 | 15 | 100 | >1000 | >1000 |
| Amphetamine | 80 * | 0.1 * | 2.5 | 60 | >1000 | >1000 | >1000 | >1000 | |
| Propranolol | 3.0/35 | 0.8/45 | >1000 | 90 | 90 | 160 | >1000 | 220 | |
| Salbutamol | 30 | 0.3 | >1000 | 180 | 0.6 | 1.0/130 | 160 | >1000 | 50 |
| Isoproterenol | 40 | 0.1 | 13 | 13 | 0.1 | 11 | 130 | >1000 | >1000 |
| Phenylephrine | 30 | 0.13 | >1000 | 20 | 17 | 20 | >1000 | >1000 | >1000 |
| Tyramine | 12 * | 50 | 210 | 90 | 60 | 80 | 200 | >1000 | 40 |
| Octopamine | 80 * | 3.2 * | >1000 | 55 | 100 | 220 | >1000 | 30 | |
| Homovanillic Acid | 80 * | >1000 * | >1000 | 85 | 1.5/90 | >1000 | >1000 | 70 | |
| Tyrosine | >1000 | >1000 | 250 | 75 | 53 | 20 | 270 | >1000 | >1000 |
| Phenylalanine | >1000 | >1000 | >1000 | 70 | 85 | >1000 | >1000 | 50 | |
| Serotonin | 45 * | 0.7 * | 45 | 60 | >1000 | 100 | >1000 | >1000 | 7 |
| Melatonin | 130 | 300 | 300 | 12 | 70 | 90 | 400 | >1000 | 150 |
| Histamine | >1000 * | >1000 * | >1000 | 210 | 70 | 110 | >1000 | >1000 | >1000 |
| Acetylcholine | 80 * | >1000 * | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| Kd (μM) @ 200 nm | Mor | Nalox | M-Enk | Epi | NorEpi | Amph | DOP | 5HT | ACh | Hist |
|---|---|---|---|---|---|---|---|---|---|---|
| MuOPR 38–51 | 35 | 0.5/35 | 0.15/55 | 1.2/35 | 1.4/45 | 1.3/90 | 60 | 100 | >1000 | >1000 |
| MuOPR 111–122 | 50 | 0.5/38 | 0.33/80 | 1.3/40 | 1.3/40 | 1.3/100 | 65 | 100 | >1000 | >1000 |
| MuOPR 121–131 | 900 | >1000 | 3.5/90 | >1000 | >1000 | >1000 | >1000 | 350 | >1000 | >1000 |
| MuOPR 132–143 | 35 | 0.5/42 | 0.4 /70 | 1.4/35 | 1.4/40 | 1.1/85 | 60 | 100 | >1000 | >1000 |
| MuOPR 211–226 | 30 | 1.0/45 | 1.0/65 | 1.2/40 | 1.3/45 | 1.2/90 | 65 | 90 | >1000 | >1000 |
| D1DR 89–98 | 20 | 5 | 80 | 400 | 300 | 530 | 75 | >1000 | >1000 | 600 |
| D1DR 170–188 | 310 | 150 | 150 | 900 | >1000 | 230 | 300 | >1000 | >1000 | >1000 |
| H1HR 77–87 | 110 | 110 | 2.3/70 | 30 | 30 | 60 | 30 | 75 | >1000 | 20 |
| B2AR 97–103 | 1 | 6 | 130 | 120 | 600 | 130 | 70 | 120 | >1000 | >1000 |
| B2AR 175–188 | 50 | 40 | 700 | 900 | 1000 | 2.3/600 | 800 | >1000 | >1000 | >1000 |
| INSR 157–166 | 60 | 200 | 100 | >1000 | 140 | 400 | >1000 | 110 | >1000 | 110 |
| INSR 281–299 | >1000 | >1000 | >1000 | 200 | >1000 | >1000 | >1000 | 900 | >1000 | >1000 |
| Kd @ 210 nm (μM) | Kd @ 200 nm (μM) | |
|---|---|---|
| Acetylcholine | >1000 | >1000 |
| Histamine | >1000 | >1000 |
| Ascorbic Acid (Vitamin C) | >1000 | >1000 |
| Naloxone | 40 | 2.5/80 |
| Methadone | 50 | 60 |
| Epinephrine | 20/1000 | 30 |
| Methionine-Enkephalin | 10 | 0.8/15 |
| Methionine-Enkephalin + Epinephrine | 4 | <0.01/4 |
| Morphine | 20 | 0.9/60 |
| Morphine + Epinephrine | 6 | <0.01/0.9/9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R.; Churchill, B.; Turke, M.; Tiruttani Subhramanyam, U.K.; Labahn, J. Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities. Int. J. Mol. Sci. 2019, 20, 4137. https://doi.org/10.3390/ijms20174137
Root-Bernstein R, Churchill B, Turke M, Tiruttani Subhramanyam UK, Labahn J. Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities. International Journal of Molecular Sciences. 2019; 20(17):4137. https://doi.org/10.3390/ijms20174137
Chicago/Turabian StyleRoot-Bernstein, Robert, Beth Churchill, Miah Turke, Udaya K. Tiruttani Subhramanyam, and Joerg Labahn. 2019. "Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities" International Journal of Molecular Sciences 20, no. 17: 4137. https://doi.org/10.3390/ijms20174137
APA StyleRoot-Bernstein, R., Churchill, B., Turke, M., Tiruttani Subhramanyam, U. K., & Labahn, J. (2019). Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities. International Journal of Molecular Sciences, 20(17), 4137. https://doi.org/10.3390/ijms20174137