Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
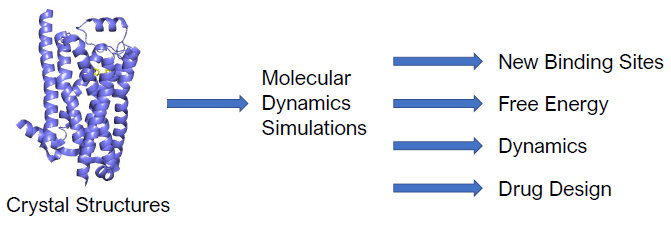
1. Introduction
2. New Insights from Molecular Dynamics Simulations
2.1. Using Molecular Dynamics Simulations to Study GPCR-Ligand Binding
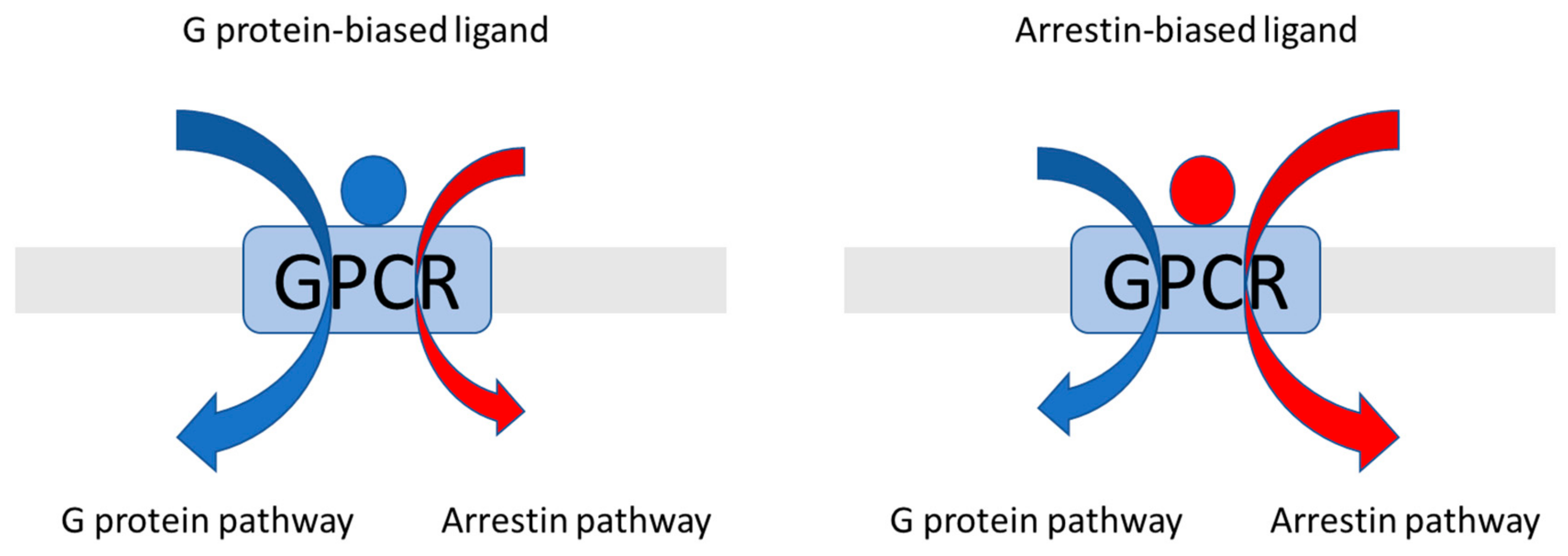
2.2. Using Molecular Dynamics to Predict Arrestin-Biased Ligands
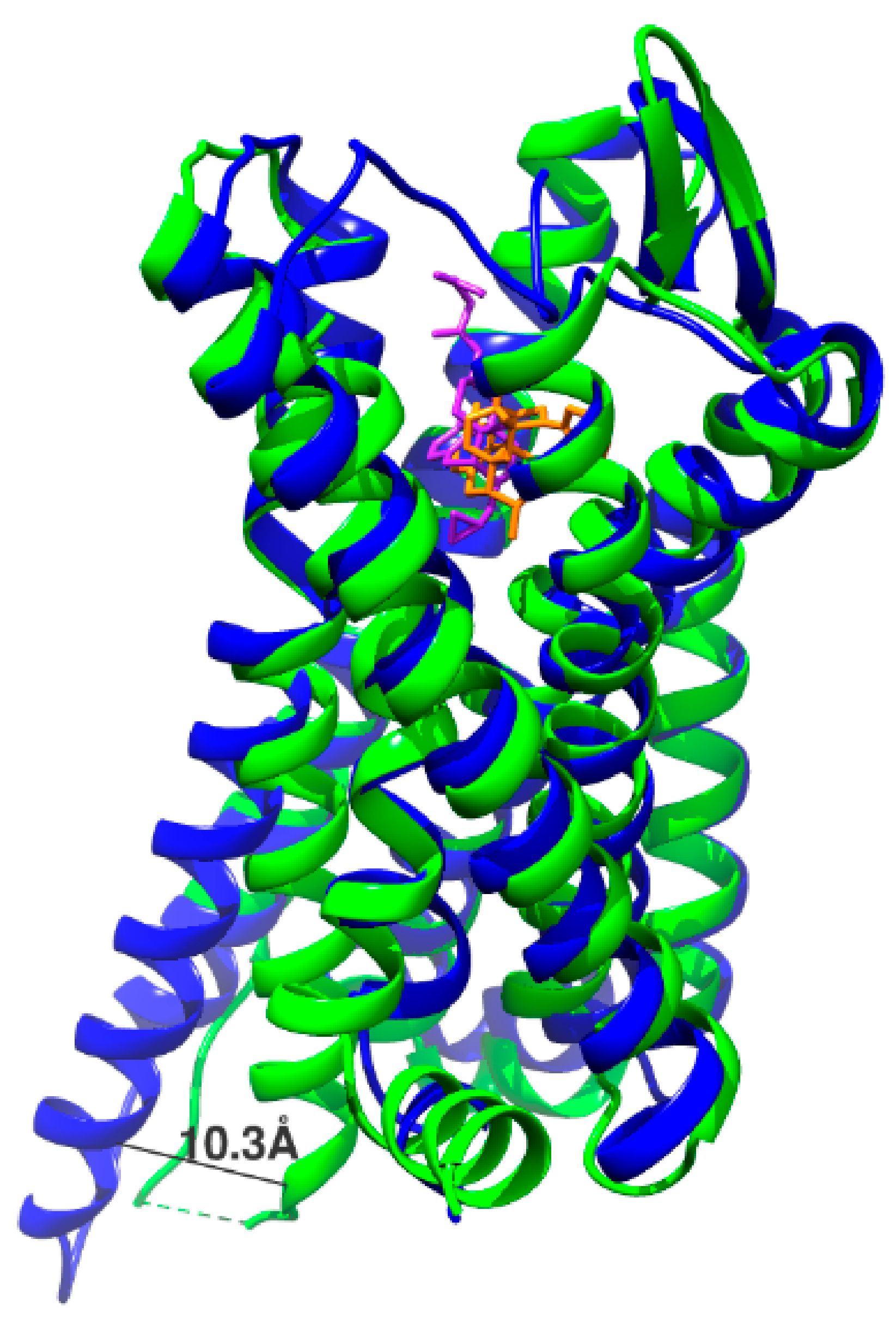
2.3. Identification of New Ligand Binding Sites and Activation Mechanisms by Accelerated Molecular Dynamics and Metadynamics Simulation
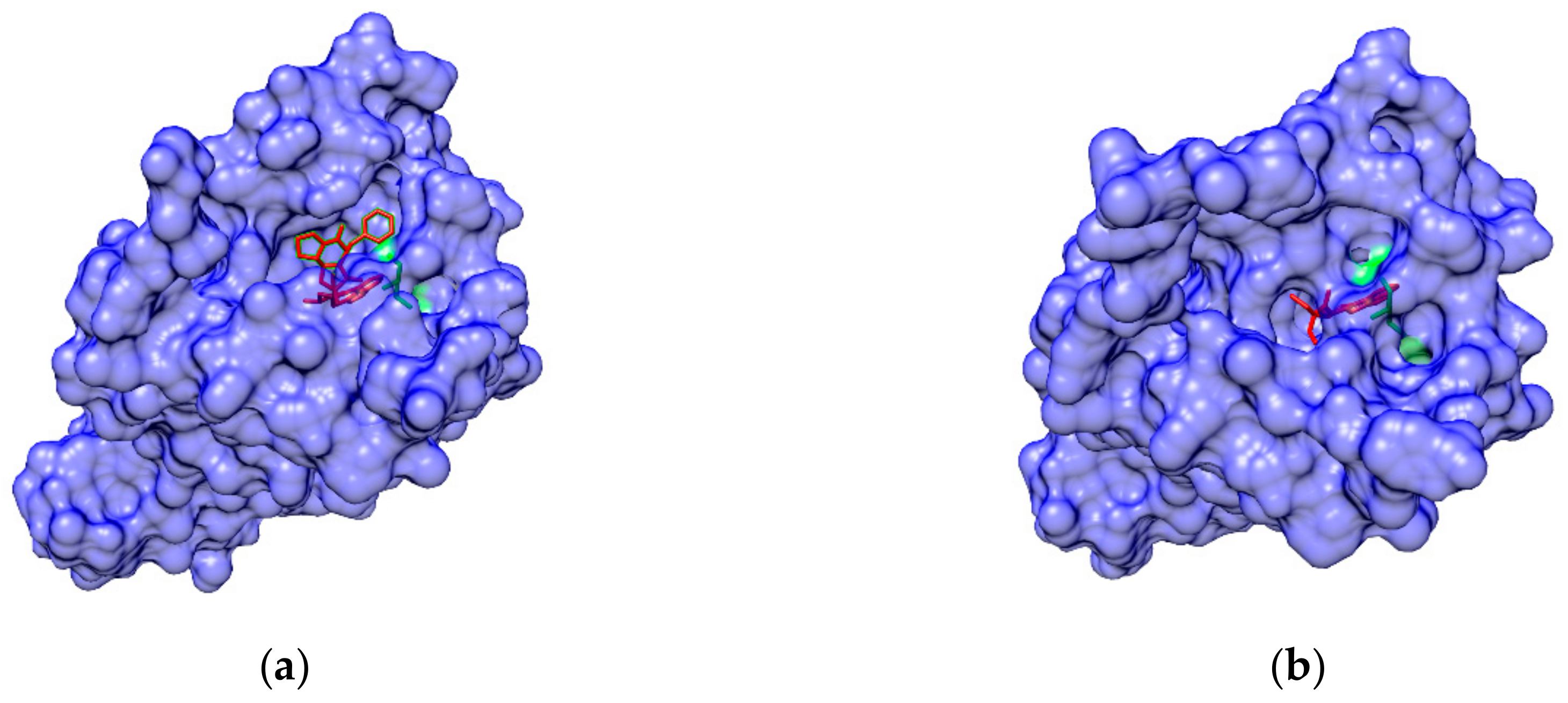
2.4. Study of Allosteric Modulation and Dynamics of GPCR-Ligand Binding
3. Insights of Molecular Dynamics to Drug Design
4. Summary and Perspectives
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| GDP | Guanosine diphosphate |
| RTKs | Receptor tyrosine kinases |
| MAP | Mitogen-activated protein |
| GRKs | GPCR kinases |
| μOR/MOR | μ-opioid receptor |
| MD | Molecular dynamics |
| cMD | Conventional molecular dynamics |
| aMD | Accelerated molecular dynamics |
| PMEMD | Particle Mesh Ewald Molecular Dynamics |
| GAFF | General AMBER force field |
| PDB | Protein Data Bank |
| ACh | Acetylcholine |
| POPC | Phosphatidylcholine |
| TTP | Tiotropium |
| SBDD | Structure-based drug design |
References
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Davenport, A.P.; Kelly, E.; Marrion, N.; Peters, J.A.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Southan, C. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. Br. J. Pharmacol. 2015, 172, 5744–5869. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Roth, B.L. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 117–144. [Google Scholar] [CrossRef]
- Roth, B.L.; Kroeze, W.K. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. J. Biol. Chem. 2015, 290, 19471–19477. [Google Scholar] [CrossRef]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef]
- Zhou, X.E.; Melcher, K.; Xu, H.E. Understanding the GPCR biased signaling through G protein and arrestin complex structures. Curr. Opin. Struct. Biol. 2017, 45, 150–159. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Lee, Y.; Lazim, R.; Macalino, S.J.Y.; Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 2019, 55, 147–153. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Rutkowska, E.; Bartuzi, D.; Targowska-Duda, K.M.; Matosiuk, D.; Selent, J. Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 2016, 132, 359–399. [Google Scholar] [PubMed]
- Schiaffino, M.V.; d’Addio, M.; Alloni, A.; Baschirotto, C.; Valetti, C.; Cortese, K.; Puri, C.; Bassi, M.T.; Colla, C.; De Luca, M. Ocular albinism: Evidence for a defect in an intracellular signal transduction system. Nat. Genet. 1999, 23, 108. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Rios, C.; Trapaidze, N.; Devi, L.A. G protein coupled receptor dimerization: Implications in modulating receptor function. J. Mol. Med. 2001, 79, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557. [Google Scholar] [CrossRef]
- Schorb, W.; Conrad, K.M.; Singer, H.A.; Dostal, D.E.; Baker, K.M. Angiotensin II is a potent stimulator of MAP-kinase activity in neonatal rat cardiac fibroblasts. J. Mol. Cell. Cardiol. 1995, 27, 1151–1160. [Google Scholar] [CrossRef]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004, 25, 413–422. [Google Scholar] [CrossRef]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S46–S55. [Google Scholar] [CrossRef]
- Scheerer, P.; Sommer, M.E. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 2017, 45, 160–169. [Google Scholar] [CrossRef]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin 2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [PubMed]
- Benovic, J.; Kühn, H.; Weyand, I.; Codina, J.; Caron, M.; Lefkowitz, R. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar] [CrossRef] [PubMed]
- Craft, C.M.; Whitmore, D.H.; Wiechmann, A.F. Cone arrestin identified by targeting expression of a functional family. J. Biol. Chem. 1994, 269, 4613–4619. [Google Scholar] [PubMed]
- Gainetdinov, R.R.; Premont, R.T.; Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004, 27, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Rajagopal, S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J. Biol. Chem. 2016, 291, 8969–8977. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of beta2-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Vaidehi, N.; Bhattacharya, S. Allosteric communication pipelines in G-protein-coupled receptors. Curr. Opin. Pharmacol. 2016, 30, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L. Structural basis for chemokine recognition and activation of a viral G protein–coupled receptor. Science 2015, 347, 1113–1117. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into mu-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101. [Google Scholar] [CrossRef]
- Yao, X.J.; Velez Ruiz, G.; Whorton, M.R.; Rasmussen, S.G.F.; DeVree, B.T.; Deupi, X.; Sunahara, R.K.; Kobilka, B. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. USA 2009, 106, 9501–9506. [Google Scholar] [CrossRef] [PubMed]
- Vaidehi, N.; Kenakin, T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr. Opin. Pharmacol. 2010, 10, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Advances in G protein-coupled receptor allostery: From function to structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Dore, A.S.; Okrasa, K.; Patel, J.C.; Serrano-Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [Google Scholar] [CrossRef]
- Jazayeri, A.; Dore, A.S.; Lamb, D.; Krishnamurthy, H.; Southall, S.M.; Baig, A.H.; Bortolato, A.; Koglin, M.; Robertson, N.J.; Errey, J.C.; et al. Extra-helical binding site of a glucagon receptor antagonist. Nature 2016, 533, 274–277. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, T.W.; Andricioaei, I.; Antonny, B.; Baum, D.; Brannigan, G.; Buchete, N.-V.; Deckman, J.T.; Delemotte, L.; Del Val, C. Membrane protein structure, function, and dynamics: A perspective from experiments and theory. J. Membr. Biol. 2015, 248, 611–640. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Khalili-Araghi, F.; Gumbart, J.; Wen, P.-C.; Sotomayor, M.; Tajkhorshid, E.; Schulten, K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol. 2009, 19, 128–137. [Google Scholar] [CrossRef]
- Stansfeld, P.J.; Sansom, M.S. Molecular simulation approaches to membrane proteins. Structure 2011, 19, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Pitman, M.C.; Feller, S.E.; Soubias, O.; Gawrisch, K. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J. Mol. Biol. 2008, 381, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Teo, I.; Roux, B.; Schulten, K. Reconciling the roles of kinetic and thermodynamic factors in membrane—protein insertion. J. Am. Chem. Soc. 2013, 135, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Marino, K.A.; Filizola, M. Investigating small-molecule ligand binding to G protein-coupled receptors with biased or unbiased molecular dynamics simulations. In Computational Methods for GPCR Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 351–364. [Google Scholar]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.A.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.S.; Hruby, V.J. Fentanyl-related compounds and derivatives: Current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014, 6, 385–412. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Bohn, L.M. Mu opioid receptor regulation and opiate responsiveness. AAPS J. 2005, 7, E587–E591. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.E.; Kelly, E. A biased view of mu opioid receptors? Mol. Pharmacol. 2019, mol. 119.115956. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the mu-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Kaserer, T.; Lantero, A.; Schmidhammer, H.; Spetea, M.; Schuster, D. Mu Opioid receptor: Novel antagonists and structural modeling. Sci. Rep. 2016, 6, 21548. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hubner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Bremer, P.T.; Kimishima, A.; Schlosburg, J.E.; Zhou, B.; Collins, K.C.; Janda, K.D. Combatting Synthetic Designer Opioids: A Conjugate Vaccine Ablates Lethal Doses of Fentanyl Class Drugs. Angew. Chem. Int. Ed. 2016, 55, 3772–3775. [Google Scholar] [CrossRef]
- Zheng, H.; Chu, J.; Zhang, Y.; Loh, H.H.; Law, P.Y. Modulating mu-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability. J. Biol. Chem. 2011, 286, 12724–12733. [Google Scholar] [CrossRef] [PubMed]
- Pil, J.; Tytgat, J. Serine 329 of the mu-opioid receptor interacts differently with agonists. J. Pharmacol. Exp. Ther. 2003, 304, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Ulens, C.; Van Boven, M.; Daenens, P.; Tytgat, J. Interaction of p-fluorofentanyl on cloned human opioid receptors and exploration of the role of Trp-318 and His-319 in μ-opioid receptor selectivity. J. Pharmacol. Exp. Ther. 2000, 294, 1024–1033. [Google Scholar] [PubMed]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lipinski, P.F.J.; Jaronczyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular dynamics of fentanyl bound to mu-opioid receptor. J. Mol. Model. 2019, 25, 144. [Google Scholar] [CrossRef]
- Marino, K.A.; Shang, Y.; Filizola, M. Insights into the function of opioid receptors from molecular dynamics simulations of available crystal structures. Br. J. Pharmacol. 2018, 175, 2834–2845. [Google Scholar] [CrossRef]
- Shang, Y.; LeRouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, K.J.; Henderson, G.; Kelly, E.; Sessions, R.B. Drug binding poses relate structure with efficacy in the μ opioid receptor. J. Mol. Biol. 2017, 429, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. PAIN 2014, 155, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- McCorvy, J.D.; Butler, K.V.; Kelly, B.; Rechsteiner, K.; Karpiak, J.; Betz, R.M.; Kormos, B.L.; Shoichet, B.K.; Dror, R.O.; Jin, J.; et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 2018, 14, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017, 168, 377–389.e12. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef]
- Esguerra, M.; Siretskiy, A.; Bello, X.; Sallander, J.; Gutiérrez-de-Terán, H. GPCR-ModSim: A comprehensive web based solution for modeling G-protein coupled receptors. Nucleic Acids Res. 2016, 44, W455–W462. [Google Scholar] [CrossRef]
- Zeng, L.; Guan, M.; Jin, H.; Liu, Z.; Zhang, L. Integrating Pharmacophore into Membrane Molecular Dynamics Simulations to Improve Homology Modeling of G Protein-coupled Receptors with Ligand Selectivity: A2A Adenosine Receptor as an Example. Chem. Biol. Drug Des. 2015, 86, 1438–1450. [Google Scholar] [CrossRef]
- Kohlhoff, K.J.; Shukla, D.; Lawrenz, M.; Bowman, G.R.; Konerding, D.E.; Belov, D.; Altman, R.B.; Pande, V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014, 6, 15. [Google Scholar] [CrossRef]
- Bai, Q.; Shi, D.; Zhang, Y.; Liu, H.; Yao, X. Exploration of the antagonist CP-376395 escape pathway for the corticotropin-releasing factor receptor 1 by random acceleration molecular dynamics simulations. Mol. Biosyst. 2014, 10, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Hernández, C.X.; Weber, J.K.; Pande, V.S. Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 2015, 48, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Vaidehi, N.; Bhattacharya, S.; Larsen, A.B. Structure and dynamics of G-protein coupled receptors. In G Protein-Coupled Receptors—Modeling and Simulation; Springer: Berlin/Heidelberg, Germany, 2014; pp. 37–54. [Google Scholar]
- Xiao, X.; Zeng, X.; Yuan, Y.; Gao, N.; Guo, Y.; Pu, X.; Li, M. Understanding the conformation transition in the activation pathway of β2 adrenergic receptor via a targeted molecular dynamics simulation. Phys. Chem. Chem. Phys. 2015, 17, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Z.; Liu, P.; Li, D.; Lin, J. An insight into antagonist binding and induced conformational dynamics of class B GPCR corticotropin-releasing factor receptor 1. Mol. Biosyst. 2015, 11, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, D.; De Graaf, C.; Moeller, A.; West, G.M.; Dharmarajan, V.; Wang, C.; Siu, F.Y.; Song, G.; Reedtz-Runge, S. Conformational states of the full-length glucagon receptor. Nat. Commun. 2015, 6, 7859. [Google Scholar] [CrossRef] [PubMed]
- Maximova, T.; Moffatt, R.; Ma, B.; Nussinov, R.; Shehu, A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Deflorian, F.; Mishra, S.; Costanzi, S. Pharmacochemistry of the platelet purinergic receptors. Purinergic Signal. 2011, 7, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, C.; Liu, P.; Li, D.; Lin, J. Identification of a Different Agonist-Binding Site and Activation Mechanism of the Human P2Y 1 Receptor. Sci. Rep. 2017, 7, 13764. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320–1335. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Felder, C.C.; Goldsmith, P.J.; Jackson, K.; Sanger, H.E.; Evans, D.A.; Mogg, A.J.; Broad, L.M. Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 2018, 136 Pt C, 449–458. [Google Scholar] [CrossRef]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Wang, J.; Palczewski, K.; Filipek, S.; Vogel, H.; Liu, Z.J.; Yuan, S. Exploring a new ligand binding site of G protein-coupled receptors. Chem. Sci. 2018, 9, 6480–6489. [Google Scholar] [CrossRef]
- Bock, A.; Schrage, R.; Mohr, K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 2018, 136 Pt C, 427–437. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Jazayeri, A.; Dias, J.M.; Marshall, F.H. From G Protein-coupled Receptor Structure Resolution to Rational Drug Design. J. Biol. Chem. 2015, 290, 19489–19495. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Leurs, R.; De Esch, I.J.; de Graaf, C. From three-dimensional GPCR structure to rational ligand discovery. In G Protein-Coupled Receptors—Modeling and Simulation; Springer: Berlin/Heidelberg, Germany, 2014; pp. 129–157. [Google Scholar]
- Kumari, P.; Ghosh, E.; Shukla, A.K. Emerging Approaches to GPCR Ligand Screening for Drug Discovery. Trends Mol. Med. 2015, 21, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kobilka, B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Armutlulu, A.; Ekesan, S.; Haliloglu, T. MCPath: Monte Carlo path generation approach to predict likely allosteric pathways and functional residues. Nucleic Acids Res. 2013, 41, W249–W255. [Google Scholar] [CrossRef] [PubMed]
- Marti-Solano, M.; Kaczor, A.A.; Guixà-González, R.; Selent, J. Computational Strategies to Incorporate GPCR Complexity in Drug Design. In Frontiers in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 3–43. [Google Scholar]
- Huang, X.-P.; Karpiak, J.; Kroeze, W.K.; Zhu, H.; Chen, X.; Moy, S.S.; Saddoris, K.A.; Nikolova, V.D.; Farrell, M.S.; Wang, S. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Soubias, O.; Teague, W.E., Jr.; Hines, K.G.; Mitchell, D.C.; Gawrisch, K. Contribution of membrane elastic energy to rhodopsin function. Biophys. J. 2010, 99, 817–824. [Google Scholar] [CrossRef]
- Zaitseva, E.; Brown, M.F.; Vogel, R. Sequential rearrangement of interhelical networks upon rhodopsin activation in membranes: The Meta II(a) conformational substate. J. Am. Chem. Soc. 2010, 132, 4815–4821. [Google Scholar] [CrossRef] [PubMed]
- Crozier, P.S.; Stevens, M.J.; Forrest, L.R.; Woolf, T.B. Molecular dynamics simulation of dark-adapted rhodopsin in an explicit membrane bilayer: Coupling between local retinal and larger scale conformational change. J. Mol. Biol. 2003, 333, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, Y.; Ewalt, J.; Ng, H.-L. Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4237. https://doi.org/10.3390/ijms20174237
Zou Y, Ewalt J, Ng H-L. Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery. International Journal of Molecular Sciences. 2019; 20(17):4237. https://doi.org/10.3390/ijms20174237
Chicago/Turabian StyleZou, Ye, John Ewalt, and Ho-Leung Ng. 2019. "Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery" International Journal of Molecular Sciences 20, no. 17: 4237. https://doi.org/10.3390/ijms20174237
APA StyleZou, Y., Ewalt, J., & Ng, H.-L. (2019). Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery. International Journal of Molecular Sciences, 20(17), 4237. https://doi.org/10.3390/ijms20174237