DNA- and DNA-Protein-Crosslink Repair in Plants


Abstract
:1. Introduction
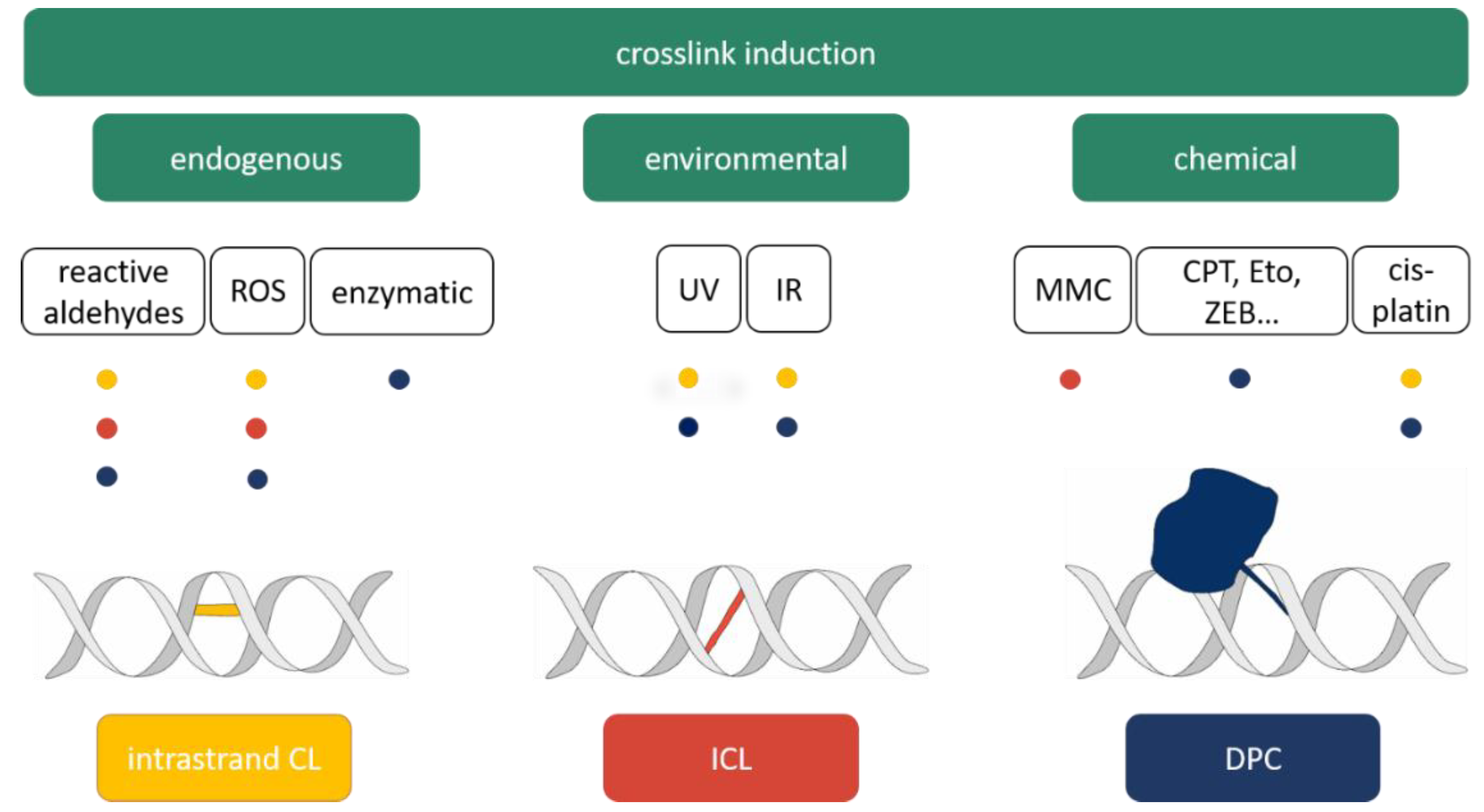
2. Sources of DNA Crosslinks
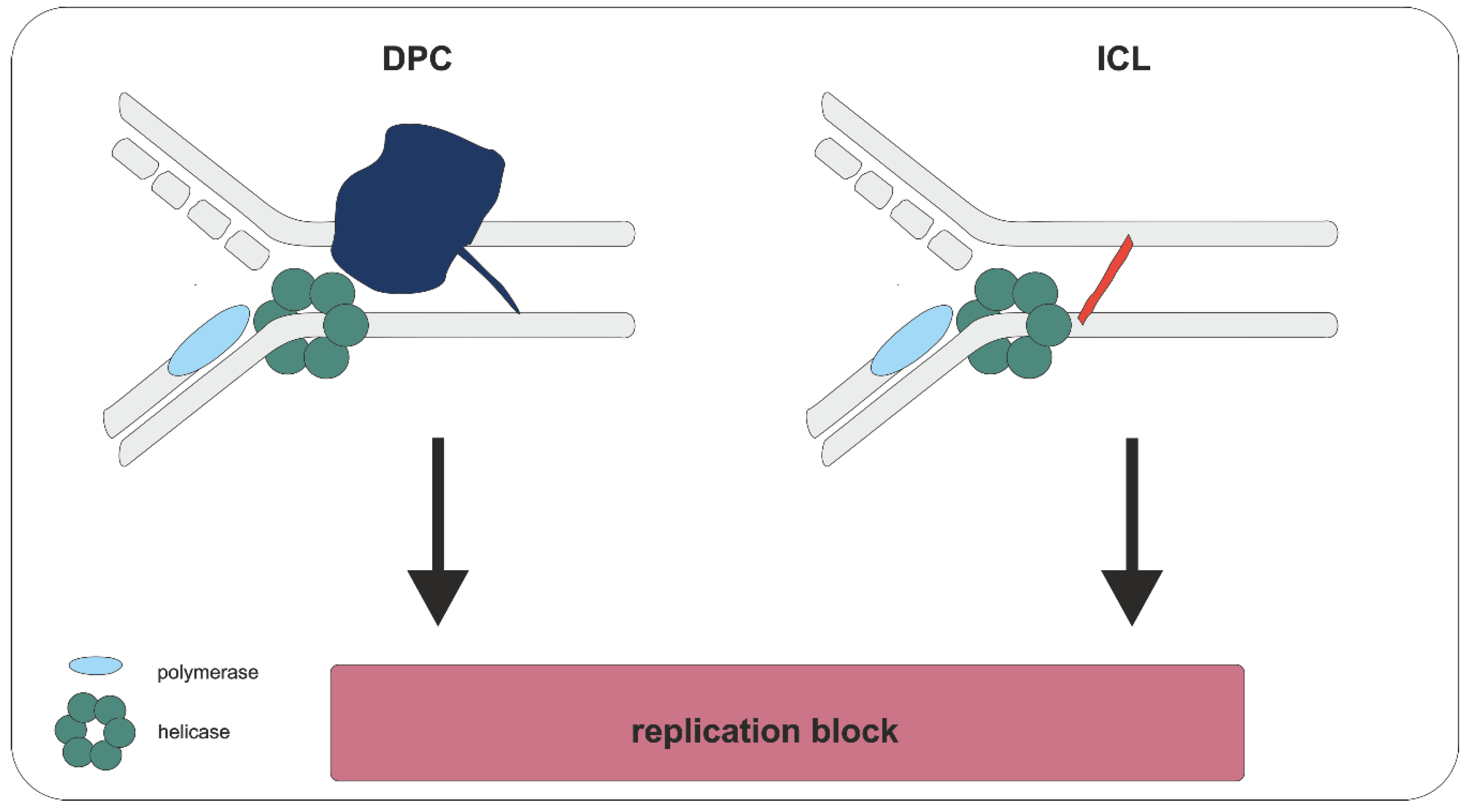
3. Biological Consequences
4. Repair of DPCs
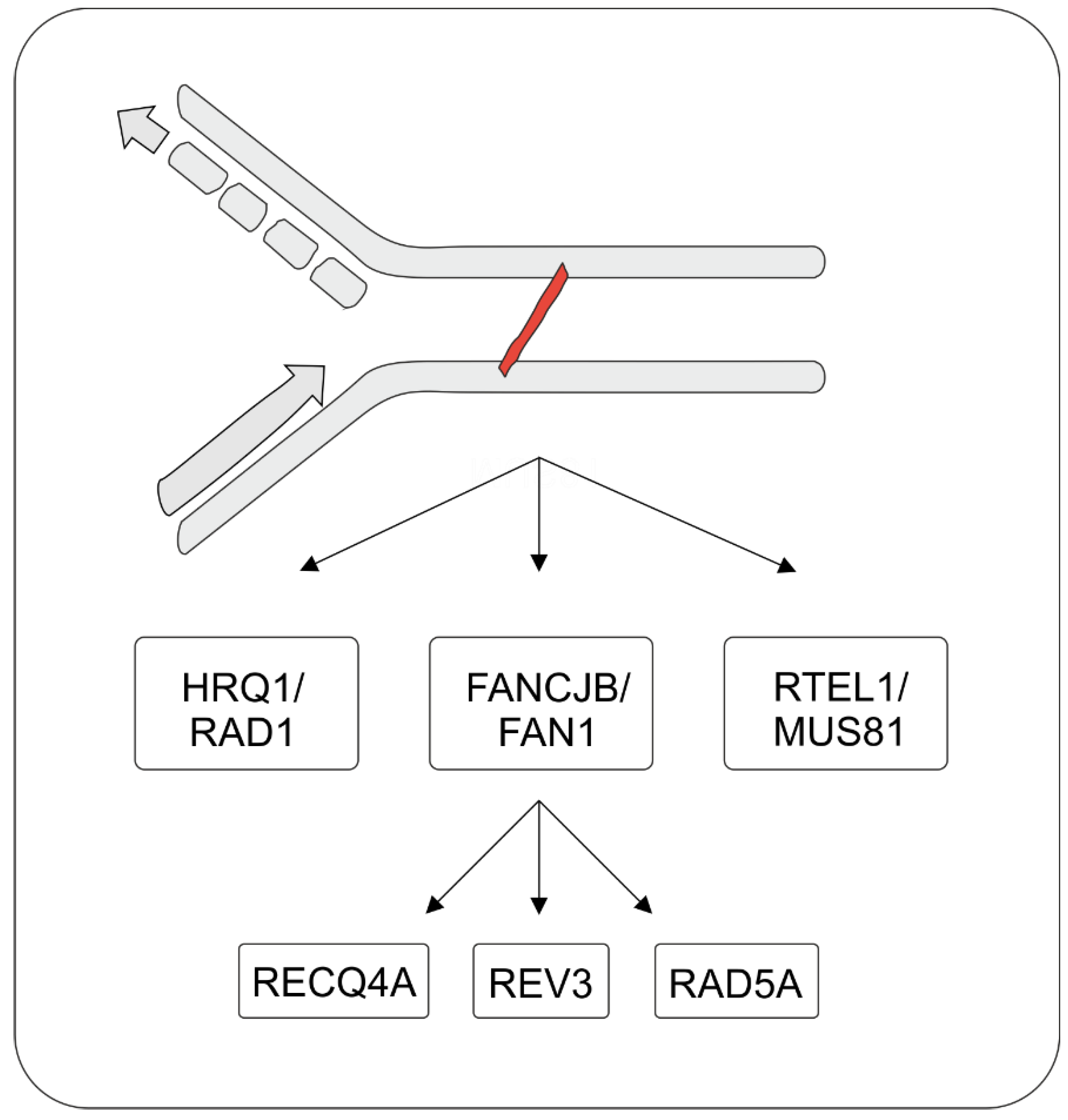
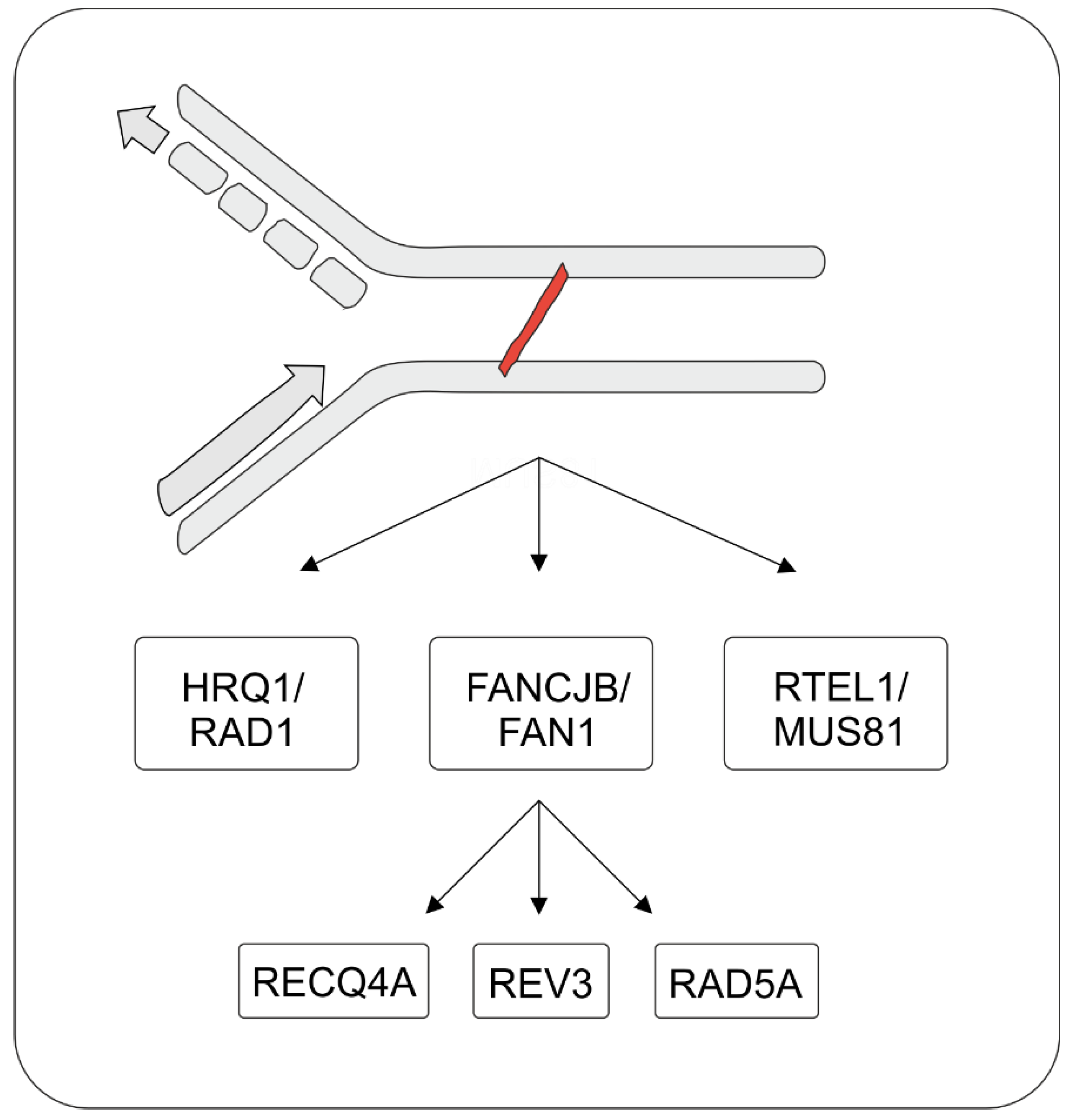
5. Repair of Intrastrand CLs and ICLs
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Base excision repair |
| CL | Crosslink |
| CPT | Camptothecin |
| DPC | DNA-protein crosslink |
| DSB | Double-strand break |
| Eto | Etoposide |
| FANC | Fanconi anemia complementation group |
| HR | Homologous recombination |
| ICL | Interstrand crosslink |
| IR | Ionizing radiation |
| MMC | Mitomycin C |
| MMEJ | Micro-homology mediated end-joining |
| NER | Nucleotide excision repair |
| PRR | Post replicative repair |
| ROS | Reactive oxygen species |
| TDP1/2 | Tyrosyl-DNA-phosphodiesterase 1/2 |
| TLS | Translesion synthesis |
| TOP1cc | Topoisomerase 1 cleavage complex |
| ZEB | Zebularine |
References
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 2011, 120 (Suppl. 1), S130–S145. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Lorenti Garcia, C.; Mechilli, M.; Proietti De Santis, L.; Schinoppi, A.; Kobos, K.; Palitti, F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat. Res. 2009, 662, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Ye, W.; Zhou, L.; Collins, L.B.; Chen, X.; Gold, A.; Ball, L.M.; Swenberg, J.A. Structural characterization of formaldehyde-induced cross-links between amino acids and deoxynucleosides and their oligomers. J. Am. Chem. Soc. 2010, 132, 3388–3399. [Google Scholar] [CrossRef] [PubMed]
- Klages-Mundt, N.L.; Li, L. Formation and repair of DNA-protein crosslink damage. Sci. China Life Sci. 2017, 60, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellarco, V.L. A mutagenicity assessment of acetaldehyde. Mutat. Res. 1988, 195, 1–20. [Google Scholar] [CrossRef]
- Chaw, Y.F.; Crane, L.E.; Lange, P.; Shapiro, R. Isolation and identification of cross-links from formaldehyde-treated nucleic acids. Biochemistry 1980, 19, 5525–5531. [Google Scholar] [CrossRef] [PubMed]
- Quievryn, G.; Zhitkovich, A. Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 2000, 21, 1573–1580. [Google Scholar] [CrossRef]
- Asada, K. Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol. 2006, 141, 391–396. [Google Scholar] [CrossRef]
- Roldán-Arjona, T.; Ariza, R.R. Repair and tolerance of oxidative DNA damage in plants. Mutat. Res. 2009, 681, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gapper, C.; Dolan, L. Control of plant development by reactive oxygen species. Plant Physiol. 2006, 141, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yao, J. Chloroplasts at the Crossroad of Photosynthesis, Pathogen Infection and Plant Defense. Int. J. Mol. Sci. 2018, 19, 3900. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.M.; Jensen, P.E.; Hansson, A. Oxidative modifications to cellular components in plants. Annu. Rev. Plant Biol. 2007, 58, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Beckhauser, T.F.; Francis-Oliveira, J.; de Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. (Lond.) 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Ham, Y.-H.; Jin, L.; Chan, H.W.; Wong, Y.-L.; Chan, C.-K.; Chung, P.-Y. Quantification of a Novel DNA-Protein Cross-Link Product Formed by Reacting Apurinic/Apyrimidinic Sites in DNA with Cysteine Residues in Protein by Liquid Chromatography-Tandem Mass Spectrometry Coupled with the Stable Isotope-Dilution Method. Anal. Chem. 2019, 91, 4987–4994. [Google Scholar] [CrossRef]
- Wang, Y. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 2008, 21, 276–281. [Google Scholar] [CrossRef]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Wang, Y.F.; Cantemir, C.; Muller, M.T. Endogenous assays of DNA methyltransferases: Evidence for differential activities of DNMT1, DNMT2, and DNMT3 in mammalian cells in vivo. Mol. Cell. Biol. 2003, 23, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Lippke, J.A.; Gordon, L.K.; Brash, D.E.; Haseltine, W.A. Distribution of UV light-induced damage in a defined sequence of human DNA: Detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6-4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Formation and processing of UV photoproducts: Effects of DNA sequence and chromatin environment. Photochem. Photobiol. 1997, 65, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Anselmino, C.; Douki, T.; Voituriez, L. Photochemistry of nucleic acids in cells. J. Photochem. Photobiol. B Biol. 1992, 15, 277–298. [Google Scholar] [CrossRef]
- Barker, S.; Weinfeld, M.; Murray, D. DNA-protein crosslinks: Their induction, repair, and biological consequences. Mutat. Res. 2005, 589, 111–135. [Google Scholar] [CrossRef]
- Nakano, T.; Xu, X.; Salem, A.M.H.; Shoulkamy, M.I.; Ide, H. Radiation-induced DNA-protein cross-links: Mechanisms and biological significance. Free Radic. Biol. Med. 2017, 107, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Stingele, J.; Habermann, B.; Jentsch, S. DNA-protein crosslink repair: Proteases as DNA repair enzymes. Trends Biochem. Sci. 2015, 40, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Hoshi, T.; Kanamori, K.; Matsumae, A.; Sano, Y.; Shima, T.; Sugawara, R. Mitomycin, a new antibiotic from Streptomyces. I. J. Antibiot. 1956, 9, 141–146. [Google Scholar] [PubMed]
- Iyer, V.N.; Szybalski, W. A molecular mechanism of mitomycin action: Linking of complementary DNA strands. Proc. Natl. Acad. Sci. USA 1963, 50, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1985, 54, 665–697. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [PubMed]
- Pommier, Y.; Huang, S.-Y.N.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst.) 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Meresse, P.; Dechaux, E.; Monneret, C.; Bertounesque, E. Etoposide: Discovery and medicinal chemistry. Curr. Med. Chem. 2004, 11, 2443–2466. [Google Scholar] [CrossRef]
- Liu, C.-H.; Finke, A.; Díaz, M.; Rozhon, W.; Poppenberger, B.; Baubec, T.; Pecinka, A. Repair of DNA Damage Induced by the Cytidine Analog Zebularine Requires ATR and ATM in Arabidopsis. Plant Cell 2015, 27, 1788–1800. [Google Scholar] [CrossRef] [Green Version]
- Kociba, R.J.; Sleight, S.D.; Rosenberg, B. Inhibition of Dunning asc itic leukemia and Walker 256 carcinosarcoma with cis-diamminedichloroplatinum (NSC-119875). Cancer Chemother. Rep. 1970, 54, 325–328. [Google Scholar] [PubMed]
- Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1986, 25, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Fichtinger-Schepman, A.M.; van der Veer, J.L.; den Hartog, J.H.; Lohman, P.H.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cis-platin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Groehler, A.; Michaelson-Richie, E.D.; Villalta, P.W.; Campbell, C.; Tretyakova, N.Y. Mass Spectrometry Based Proteomics Study of Cis-platin-Induced DNA-Protein Cross-Linking in Human Fibrosarcoma (HT1080) Cells. Chem. Res. Toxicol. 2017, 30, 980–995. [Google Scholar] [CrossRef] [PubMed]
- Zwelling, L.A.; Anderson, T.; Kohn, K.W. DNA-protein and DNA interstrand cross-linking by cis- and trans-platinum(II) diamminedichloride in L1210 mouse leukemia cells and relation to cytotoxicity. Cancer Res. 1979, 39, 365–369. [Google Scholar] [PubMed]
- Woźniak, K.; Walter, Z. Induction of DNA-protein cross-links by platinum compounds. Z. Naturforsch. C J. Biosci. 2000, 55, 731–736. [Google Scholar] [CrossRef]
- Olinski, R.; Wedrychowski, A.; Schmidt, W.N.; Briggs, R.C.; Hnilica, L.S. In vivo DNA-protein cross-linking by cis- and trans-diamminedichloroplatinum(II). Cancer Res. 1987, 47, 201–205. [Google Scholar]
- Chválová, K.; Brabec, V.; Kaspárková, J. Mechanism of the formation of DNA-protein cross-links by antitumor cis-platin. Nucleic Acids Res. 2007, 35, 1812–1821. [Google Scholar] [CrossRef]
- Tretyakova, N.Y.; Groehler, A.; Ji, S. DNA-Protein Cross-Links: Formation, Structural Identities, and Biological Outcomes. Acc. Chem. Res. 2015, 48, 1631–1644. [Google Scholar] [CrossRef]
- Lewis, J.S.; Jergic, S.; Dixon, N.E. The E. coli DNA Replication Fork. Enzymes 2016, 39, 31–88. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Richardson, C.C. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009, 78, 205–243. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingele, J.; Jentsch, S. DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2015, 16, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Nakano, T.; Salem, A.M.H.; Shoulkamy, M.I. DNA-protein cross-links: Formidable challenges to maintaining genome integrity. DNA Repair (Amst.) 2018, 71, 190–197. [Google Scholar] [CrossRef]
- Nakano, T.; Miyamoto-Matsubara, M.; Shoulkamy, M.I.; Salem, A.M.H.; Pack, S.P.; Ishimi, Y.; Ide, H. Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J. Biol. Chem. 2013, 288, 4649–4658. [Google Scholar] [CrossRef]
- Fu, Y.V.; Yardimci, H.; Long, D.T.; Ho, T.V.; Guainazzi, A.; Bermudez, V.P.; Hurwitz, J.; van Oijen, A.; Schärer, O.D.; Walter, J.C. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 2011, 146, 931–941. [Google Scholar] [CrossRef]
- Kuo, H.K.; Griffith, J.D.; Kreuzer, K.N. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 2007, 67, 8248–8254. [Google Scholar] [CrossRef]
- Novakova, O.; Kasparkova, J.; Malina, J.; Natile, G.; Brabec, V. DNA-protein cross-linking by trans-PtCl(2)(E-iminoether)(2). A concept for activation of the trans geometry in platinum antitumor complexes. Nucleic Acids Res. 2003, 31, 6450–6460. [Google Scholar] [CrossRef]
- Yeo, J.E.; Wickramaratne, S.; Khatwani, S.; Wang, Y.-C.; Vervacke, J.; Distefano, M.D.; Tretyakova, N.Y. Synthesis of site-specific DNA-protein conjugates and their effects on DNA replication. ACS Chem. Biol. 2014, 9, 1860–1868. [Google Scholar] [CrossRef]
- McCabe, K.M.; Olson, S.B.; Moses, R.E. DNA interstrand crosslink repair in mammalian cells. J. Cell. Physiol. 2009, 220, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Rycenga, H.B.; Long, D.T. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol. 2018, 41, 20–26. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Modesti, M.; Maas, A.; Wyman, C.; Essers, J.; Kanaar, R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006, 25, 4921–4932. [Google Scholar] [CrossRef]
- Zhu, G.; Song, L.; Lippard, S.J. Visualizing inhibition of nucleosome mobility and transcription by cis-platin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 2013, 73, 4451–4460. [Google Scholar] [CrossRef]
- Nakano, T.; Ouchi, R.; Kawazoe, J.; Pack, S.P.; Makino, K.; Ide, H. T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription. J. Biol. Chem. 2012, 287, 6562–6572. [Google Scholar] [CrossRef]
- Liu, C.; Pouliot, J.J.; Nash, H.A. Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proc. Natl. Acad. Sci. USA 2002, 99, 14970–14975. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-W.; Regairaz, M.; Seiler, J.A.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef]
- Nakano, T.; Morishita, S.; Katafuchi, A.; Matsubara, M.; Horikawa, Y.; Terato, H.; Salem, A.M.H.; Izumi, S.; Pack, S.P.; Makino, K.; et al. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Mol. Cell 2007, 28, 147–158. [Google Scholar] [CrossRef]
- Nakano, T.; Katafuchi, A.; Matsubara, M.; Terato, H.; Tsuboi, T.; Masuda, T.; Tatsumoto, T.; Pack, S.P.; Makino, K.; Croteau, D.L.; et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009, 284, 27065–27076. [Google Scholar] [CrossRef]
- De Graaf, B.; Clore, A.; McCullough, A.K. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst.) 2009, 8, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Stingele, J.; Schwarz, M.S.; Bloemeke, N.; Wolf, P.G.; Jentsch, S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 2014, 158, 327–338. [Google Scholar] [CrossRef]
- Baker, D.J.; Wuenschell, G.; Xia, L.; Termini, J.; Bates, S.E.; Riggs, A.D.; O’Connor, T.R. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 2007, 282, 22592–22604. [Google Scholar] [CrossRef]
- Orta, M.L.; Calderón-Montaño, J.M.; Domínguez, I.; Pastor, N.; Burgos-Morón, E.; López-Lázaro, M.; Cortés, F.; Mateos, S.; Helleday, T. 5-Aza-2’-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Ridpath, J.R.; Nakamura, A.; Tano, K.; Luke, A.M.; Sonoda, E.; Arakawa, H.; Buerstedde, J.-M.; Gillespie, D.A.F.; Sale, J.E.; Yamazoe, M.; et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007, 67, 11117–11122. [Google Scholar] [CrossRef]
- Sutherland, J.H.; Holloman, W.K. Loss of Cohesin Subunit Rec8 Switches Rad51 Mediator Dependence in Resistance to Formaldehyde Toxicity in Ustilago maydis. Genetics 2018, 210, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Nitiss, J.L. DNA repair functions that control sensitivity to topoisomerase-targeting drugs. Eukaryotic Cell 2004, 3, 82–90. [Google Scholar] [CrossRef]
- Woodworth, D.L.; Kreuzer, K.N. Bacteriophage T4 mutants hypersensitive to an antitumor agent that induces topoisomerase-DNA cleavage complexes. Genetics 1996, 143, 1081–1090. [Google Scholar]
- Stohr, B.A.; Kreuzer, K.N. Repair of topoisomerase-mediated DNA damage in bacteriophage T4. Genetics 2001, 158, 19–28. [Google Scholar]
- Koster, D.A.; Palle, K.; Bot, E.S.M.; Bjornsti, M.-A.; Dekker, N.H. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 2007, 448, 213–217. [Google Scholar] [CrossRef]
- Imamura, O.; Fujita, K.; Itoh, C.; Takeda, S.; Furuichi, Y.; Matsumoto, T. Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene 2002, 21, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.A.; Fan, A.M.; Meng, L.; Doe, C.F.; North, P.S.; Hickson, I.D.; Pommier, Y. Phosphorylation of BLM, dissociation from topoisomerase IIIalpha, and colocalization with gamma-H2AX after topoisomerase I-induced replication damage. Mol. Cell. Biol. 2005, 25, 8925–8937. [Google Scholar] [CrossRef]
- Hartung, F.; Suer, S.; Knoll, A.; Wurz-Wildersinn, R.; Puchta, H. Topoisomerase 3alpha and RMI1 suppress somatic crossovers and are essential for resolution of meiotic recombination intermediates in Arabidopsis thaliana. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Pouliot, J.J.; Yao, K.C.; Robertson, C.A.; Nash, H.A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 1999, 286, 552–555. [Google Scholar] [CrossRef]
- Yang, D.; Wang, A.H. Structural studies of interactions between anticancer platinum drugs and DNA. Prog. Biophys. Mol. Biol. 1996, 66, 81–111. [Google Scholar] [CrossRef]
- Interthal, H.; Pouliot, J.J.; Champoux, J.J. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. USA 2001, 98, 12009–12014. [Google Scholar] [CrossRef] [Green Version]
- Debéthune, L.; Kohlhagen, G.; Grandas, A.; Pommier, Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002, 30, 1198–1204. [Google Scholar] [CrossRef]
- Lin, C.-P.; Ban, Y.; Lyu, Y.L.; Desai, S.D.; Liu, L.F. A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J. Biol. Chem. 2008, 283, 21074–21083. [Google Scholar] [CrossRef]
- Interthal, H.; Champoux, J.J. Effects of DNA and protein size on substrate cleavage by human tyrosyl-DNA phosphodiesterase 1. Biochem. J. 2011, 436, 559–566. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.F. To live or to die: A matter of processing damaged DNA termini in neurons. EMBO Mol. Med. 2011, 3, 78–88. [Google Scholar] [CrossRef]
- Das, B.B.; Huang, S.-y.N.; Murai, J.; Rehman, I.; Amé, J.-C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.J.R.; Chiang, S.-C.; Wells, O.S.; Rookyard, C.; El-Khamisy, S.F. SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Liao, Z.Y.; Barceló, J.M.; Kohlhagen, G.; Caldecott, K.W.; Weinfeld, M.; Pommier, Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst.) 2003, 2, 1087–1100. [Google Scholar] [CrossRef]
- Pouliot, J.J.; Robertson, C.A.; Nash, H.A. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells 2001, 6, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.R.; Wilson, T.E. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl. Acad. Sci. USA 2002, 99, 13669–13674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.M.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef]
- Alagoz, M.; Wells, O.S.; El-Khamisy, S.F. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2014, 42, 3089–3103. [Google Scholar] [CrossRef]
- Gao, R.; Schellenberg, M.J.; Huang, S.-y.N.; Abdelmalak, M.; Marchand, C.; Nitiss, K.C.; Nitiss, J.L.; Williams, R.S.; Pommier, Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2·DNA and Top2·RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 2014, 289, 17960–17969. [Google Scholar] [CrossRef]
- Zeng, Z.; Cortés-Ledesma, F.; El Khamisy, S.F.; Caldecott, K.W. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2011, 286, 403–409. [Google Scholar] [CrossRef]
- Faè, M.; Balestrazzi, A.; Confalonieri, M.; Donà, M.; Macovei, A.; Valassi, A.; Giraffa, G.; Carbonera, D. Copper-mediated genotoxic stress is attenuated by the overexpression of the DNA repair gene MtTdp2α (tyrosyl-DNA phosphodiesterase 2) in Medicago truncatula plants. Plant Cell Rep. 2014, 33, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Biggins, S.; Bhalla, N.; Chang, A.; Smith, D.L.; Murray, A.W. Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 2001, 159, 453–470. [Google Scholar] [PubMed]
- Mullen, J.R.; Chen, C.-F.; Brill, S.J. Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol. Cell. Biol. 2010, 30, 3737–3748. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.R.; Das, M.; Brill, S.J. Genetic evidence that polysumoylation bypasses the need for a SUMO-targeted Ub ligase. Genetics 2011, 187, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Vaz, B.; Halder, S.; Lockhart, P.J.; Marinovic-Terzic, I.; Lopez-Mosqueda, J.; Philipp, M.; Sim, J.C.H.; Smith, K.R.; Oehler, J.; et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 2014, 46, 1239–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruijs, M.W.G.; van Andel, R.N.J.; Oshima, J.; Madan, K.; Nieuwint, A.W.M.; Aalfs, C.M. Atypical progeroid syndrome: An unknown helicase gene defect? Am. J. Med. Genet. 2003, 116, 295–299. [Google Scholar] [CrossRef]
- Stingele, J.; Bellelli, R.; Alte, F.; Hewitt, G.; Sarek, G.; Maslen, S.L.; Tsutakawa, S.E.; Borg, A.; Kjær, S.; Tainer, J.A.; et al. Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 2016, 64, 688–703. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Mosqueda, J.; Maddi, K.; Prgomet, S.; Kalayil, S.; Marinovic-Terzic, I.; Terzic, J.; Dikic, I. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife 2016, 5. [Google Scholar] [CrossRef]
- Enderle, J.; Dorn, A.; Beying, N.; Trapp, O.; Puchta, H. The Protease WSS1A, the Endonuclease MUS81, and the Phosphodiesterase TDP1 Are Involved in Independent Pathways of DNA-protein Crosslink Repair in Plants. Plant Cell 2019, 31, 775–790. [Google Scholar] [CrossRef]
- Hartung, F.; Suer, S.; Bergmann, T.; Puchta, H. The role of AtMUS81 in DNA repair and its genetic interaction with the helicase AtRecQ4A. Nucleic Acids Res. 2006, 34, 4438–4448. [Google Scholar] [CrossRef]
- Geuting, V.; Kobbe, D.; Hartung, F.; Dürr, J.; Focke, M.; Puchta, H. Two distinct MUS81-EME1 complexes from Arabidopsis process Holliday junctions. Plant Physiol. 2009, 150, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Fidantsef, A.L.; Mitchell, D.L.; Britt, A.B. The Arabidopsis UVH1 gene is a homolog of the yeast repair endonuclease RAD1. Plant Physiol. 2000, 124, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Mannuß, A.; Kobbe, D.; Knoll, A.; Trapp, O.; Dorn, A.; Puchta, H. The DNA translocase RAD5A acts independently of the other main DNA repair pathways, and requires both its ATPase and RING domain for activity in Arabidopsis thaliana. Plant J. 2017, 91, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, L.; Zhong, D. Photolyase: Dynamics and Mechanisms of Repair of Sun-Induced DNA Damage. Photochem. Photobiol. 2017, 93, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Nepal, M.; Che, R.; Ma, C.; Zhang, J.; Fei, P. FANCD2 and DNA Damage. Int. J. Mol. Sci. 2017, 18, 1804. [Google Scholar] [CrossRef] [PubMed]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Maung, K.Z.Y.; Leo, P.J.; Bassal, M.; Casolari, D.A.; Gray, J.X.; Bray, S.C.; Pederson, S.; Singhal, D.; Samaraweera, S.E.; Nguyen, T.; et al. Rare variants in Fanconi anemia genes are enriched in acute myeloid leukemia. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef]
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef]
- Huang, M.; Kim, J.M.; Shiotani, B.; Yang, K.; Zou, L.; D’Andrea, A.D. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell 2010, 39, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, L. DNA crosslinking damage and cancer—a tale of friend and foe. Transl. Cancer Res. 2013, 2, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genomics 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Knoll, A.; Higgins, J.D.; Seeliger, K.; Reha, S.J.; Dangel, N.J.; Bauknecht, M.; Schröpfer, S.; Franklin, F.C.H.; Puchta, H. The Fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell 2012, 24, 1448–1464. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.; Feller, L.; Castri, D.; Röhrig, S.; Enderle, J.; Herrmann, N.J.; Block-Schmidt, A.; Trapp, O.; Köhler, L.; Puchta, H. An Arabidopsis FANCJ helicase homologue is required for DNA crosslink repair and rDNA repeat stability. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.H. (Ed.) Molecular Biology; Springer: New York, NY, USA, 2014; ISBN 978-1-4614-7570-5. [Google Scholar]
- Dangel, N.J.; Knoll, A.; Puchta, H. MHF1 plays Fanconi anaemia complementation group M protein (FANCM)-dependent and FANCM-independent roles in DNA repair and homologous recombination in plants. Plant J. 2014, 78, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Siaud, N.; Dray, E.; Gy, I.; Gérard, E.; Takvorian, N.; Doutriaux, M.-P. Brca2 is involved in meiosis in Arabidopsis thaliana as suggested by its interaction with Dmc1. EMBO J. 2004, 23, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, K.; Dukowic-Schulze, S.; Wurz-Wildersinn, R.; Pacher, M.; Puchta, H. BRCA2 is a mediator of RAD51- and DMC1-facilitated homologous recombination in Arabidopsis thaliana. New Phytol. 2012, 193, 364–375. [Google Scholar] [CrossRef]
- Kurzbauer, M.-T.; Pradillo, M.; Kerzendorfer, C.; Sims, J.; Ladurner, R.; Oliver, C.; Janisiw, M.P.; Mosiolek, M.; Schweizer, D.; Copenhaver, G.P.; et al. Arabidopsis thaliana FANCD2 Promotes Meiotic Crossover Formation. Plant Cell 2018, 30, 415–428. [Google Scholar] [CrossRef]
- Girard, C.; Crismani, W.; Froger, N.; Mazel, J.; Lemhemdi, A.; Horlow, C.; Mercier, R. FANCM-associated proteins MHF1 and MHF2, but not the other Fanconi anemia factors, limit meiotic crossovers. Nucleic Acids Res. 2014, 42, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Yan, Z.; Wang, W. FANCL replaces BRCA1 as the likely ubiquitin ligase responsible for FANCD2 monoubiquitination. Cell Cycle 2004, 3, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Crismani, W.; Girard, C.; Froger, N.; Pradillo, M.; Santos, J.L.; Chelysheva, L.; Copenhaver, G.P.; Horlow, C.; Mercier, R. FANCM limits meiotic crossovers. Science 2012, 336, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Osakabe, K.; Nakayama, S.; Endo, M.; Tagiri, A.; Todoriki, S.; Ichikawa, H.; Toki, S. Arabidopsis RAD51C gene is important for homologous recombination in meiosis and mitosis. Plant Physiol. 2005, 139, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Dubest, S.; Gallego, M.E.; White, C.I. Role of the AtRad1p endonuclease in homologous recombination in plants. EMBO Rep. 2002, 3, 1049–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, R.; Armstrong, S.J.; Horlow, C.; Jackson, N.P.; Makaroff, C.A.; Vezon, D.; Pelletier, G.; Jones, G.H.; Franklin, F.C.H. The meiotic protein SWI1 is required for axial element formation and recombination initiation in Arabidopsis. Development 2003, 130, 3309–3318. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Herrmann, N.J.; Knoll, A.; Puchta, H. The nuclease FAN1 is involved in DNA crosslink repair in Arabidopsis thaliana independently of the nuclease MUS81. Nucleic Acids Res. 2015, 43, 3653–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, K.; Schöpf, B.; Kaden, S.; Sendoel, A.; Eberhard, R.; Lademann, C.; Cannavó, E.; Sartori, A.A.; Hengartner, M.O.; Jiricny, J. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 2010, 142, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Vannier, J.-B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.R.; Ding, H.; Boulton, S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of murine telomere length by Rtel: An essential gene encoding a helicase-like protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Youds, J.L.; Ward, J.D.; McIlwraith, M.J.; O’Neil, N.J.; Petalcorin, M.I.R.; Martin, J.S.; Collis, S.J.; Cantor, S.B.; Auclair, M.; et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 2008, 135, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Recker, J.; Knoll, A.; Puchta, H. The Arabidopsis thaliana homolog of the helicase RTEL1 plays multiple roles in preserving genome stability. Plant Cell 2014, 26, 4889–4902. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Cools, T.; Kalhorzadeh, P.; Heyman, J.; de Veylder, L. Deficiency of the Arabidopsis helicase RTEL1 triggers a SOG1-dependent replication checkpoint in response to DNA cross-links. Plant Cell 2015, 27, 149–161. [Google Scholar] [CrossRef]
- Röhrig, S.; Schröpfer, S.; Knoll, A.; Puchta, H. The RTR Complex Partner RMI2 and the DNA Helicase RTEL1 Are Both Independently Involved in Preserving the Stability of 45S rDNA Repeats in Arabidopsis thaliana. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Gallego, F.; Fleck, O.; Li, A.; Wyrzykowska, J.; Tinland, B. AtRAD1, a plant homologue of human and yeast nucleotide excision repair endonucleases, is involved in dark repair of UV damages and recombination. Plant J. 2000, 21, 507–518. [Google Scholar] [CrossRef]
- Roy, S.; Choudhury, S.R.; Sengupta, D.N.; Das, K.P. Involvement of AtPolλ in the repair of high salt- and DNA cross-linking agent-induced double strand breaks in Arabidopsis. Plant Physiol. 2013, 162, 1195–1210. [Google Scholar] [CrossRef]
- Reidt, W.; Wurz, R.; Wanieck, K.; Chu, H.H.; Puchta, H. A homologue of the breast cancer-associated gene BARD1 is involved in DNA repair in plants. EMBO J. 2006, 25, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Block-Schmidt, A.S.; Dukowic-Schulze, S.; Wanieck, K.; Reidt, W.; Puchta, H. BRCC36A is epistatic to BRCA1 in DNA crosslink repair and homologous recombination in Arabidopsis thaliana. Nucleic Acids Res. 2011, 39, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Bleuyard, J.-Y.; Gallego, M.E.; Savigny, F.; White, C.I. Differing requirements for the Arabidopsis Rad51 paralogs in meiosis and DNA repair. Plant J. 2005, 41, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Chang, Y.; Li, X.; Xiao, J.; Wang, S. The rice RAD51C gene is required for the meiosis of both female and male gametocytes and the DNA repair of somatic cells. J. Exp. Bot. 2012, 63, 5323–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannuss, A.; Dukowic-Schulze, S.; Suer, S.; Hartung, F.; Pacher, M.; Puchta, H. RAD5A, RECQ4A, and MUS81 have specific functions in homologous recombination and define different pathways of DNA repair in Arabidopsis thaliana. Plant Cell 2010, 22, 3318–3330. [Google Scholar] [CrossRef] [PubMed]
- Kobbe, S.; Trapp, O.; Knoll, A.; Manuss, A.; Puchta, H. The Translesion Polymerase ζ Has Roles Dependent on and Independent of the Nuclease MUS81 and the Helicase RECQ4A in DNA Damage Repair in Arabidopsis. Plant Physiol. 2015, 169, 2718–2729. [Google Scholar] [CrossRef]
- Hartung, F.; Suer, S.; Puchta, H. Two closely related RecQ helicases have antagonistic roles in homologous recombination and DNA repair in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 18836–18841. [Google Scholar] [CrossRef]
- Dorn, A.; Röhrig, S.; Papp, K.; Schröpfer, S.; Hartung, F.; Knoll, A.; Puchta, H. The topoisomerase 3α zinc-finger domain T1 of Arabidopsis thaliana is required for targeting the enzyme activity to Holliday junction-like DNA repair intermediates. PLoS Genet. 2018, 14, e1007674. [Google Scholar] [CrossRef]
- Röhrig, S.; Dorn, A.; Enderle, J.; Schindele, A.; Herrmann, N.J.; Knoll, A.; Puchta, H. The RecQ-like helicase HRQ1 is involved in DNA crosslink repair in Arabidopsis in a common pathway with the Fanconi anemia-associated nuclease FAN1 and the postreplicative repair ATPase RAD5A. New Phytol. 2018, 218, 1478–1490. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, G.; van Gessel, N.; Köchl, F.; Hunn, L.; Schulze, K.; Maloukh, L.; Nogué, F.; Decker, E.L.; Hartung, F.; Reski, R. RecQ Helicases Function in Development, DNA Repair, and Gene Targeting in Physcomitrella patens. Plant Cell 2018, 30, 717–736. [Google Scholar] [CrossRef]
- Osakabe, K.; Abe, K.; Yamanouchi, H.; Takyuu, T.; Yoshioka, T.; Ito, Y.; Kato, T.; Tabata, S.; Kurei, S.; Yoshioka, Y.; et al. Arabidopsis Rad51B is important for double-strand DNA breaks repair in somatic cells. Plant Mol. Biol. 2005, 57, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, K.; Abe, K.; Yoshioka, T.; Osakabe, Y.; Todoriki, S.; Ichikawa, H.; Hohn, B.; Toki, S. Isolation and characterization of the RAD54 gene from Arabidopsis thaliana. Plant J. 2006, 48, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Pradillo, M.; López, E.; Linacero, R.; Romero, C.; Cuñado, N.; Sánchez-Morán, E.; Santos, J.L. Together yes, but not coupled: New insights into the roles of RAD51 and DMC1 in plant meiotic recombination. Plant J. 2012, 69, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-P.; Mannuss, A.; Orel, N.; Heitzeberg, F.; Puchta, H. A homolog of ScRAD5 is involved in DNA repair and homologous recombination in Arabidopsis. Plant Physiol. 2008, 146, 1786–1796. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FANC Gene | Synonym | Keywords | Reference |
|---|---|---|---|
| FANCD1 | BRCA2 | Somatic and meiotic HR | [132,133] |
| FANCD2 | Somatic and meiotic HR | [134] | |
| FANCE | No role in meiotic HR | [135] | |
| FANCI | No role in meiotic HR | [135] | |
| FANCJ | BRIP1, BACH1 | Helicase, ICL repair, replicative repair, rDNA stability | [129] |
| FANCL | Ubiquitin ligase, no role in meiotic HR | [135,136] | |
| FANCM | Helicase, Antirecombinase, ICL repair, suppression of somatic and meiotic HR | [128,137] | |
| FANCO | RAD51C | Recombinase, Mitotic and meiotic HR | [138] |
| FANCQ | ERCC4, RAD1, UVH1 | Endonuclease, NER | [112,139] |
| FANCR | RAD51 | Recombinase, mitotic and meiotic HR | [133,140] |
| FANCT | uncharacterized | [127] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enderle, J.; Dorn, A.; Puchta, H. DNA- and DNA-Protein-Crosslink Repair in Plants. Int. J. Mol. Sci. 2019, 20, 4304. https://doi.org/10.3390/ijms20174304
Enderle J, Dorn A, Puchta H. DNA- and DNA-Protein-Crosslink Repair in Plants. International Journal of Molecular Sciences. 2019; 20(17):4304. https://doi.org/10.3390/ijms20174304
Chicago/Turabian StyleEnderle, Janina, Annika Dorn, and Holger Puchta. 2019. "DNA- and DNA-Protein-Crosslink Repair in Plants" International Journal of Molecular Sciences 20, no. 17: 4304. https://doi.org/10.3390/ijms20174304
APA StyleEnderle, J., Dorn, A., & Puchta, H. (2019). DNA- and DNA-Protein-Crosslink Repair in Plants. International Journal of Molecular Sciences, 20(17), 4304. https://doi.org/10.3390/ijms20174304