Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article

Abstract
:1. Introduction
2. Important Characteristics and Mechanisms of the Pulmonary Endothelium
3. Inflammatory Cells and Mediators in COPD
3.1. Neutrophils’ Migration and Netosis
3.2. Macrophages and Eosinophils
3.3. Inflammatory Mediators
4. Endothelial Dysfunction and Apoptosis
5. Dysfunction of the Endothelium and its Effects in COPD Patients: Pulmonary Hypertension
6. Smoking and Endothelial Dysfunction
7. Arterial Stiffness, Aortic Pulse wave Velocity and Pulmonary Rehabilitation
8. Conclusion and Future Perspectives
Funding
Conflicts of Interest
References
- Spruit, M.A.; Pennings, H.J.; Janssen, P.P.; Does, J.D.; Scroyen, S.; Akkermans, M.A.; Mostert, R.; Wouters, E.F. Extra-pulmonary features in COPD patients entering rehabilitation after stratification for MRC dyspnea grade. Respir. Med. 2007, 101, 2454–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J.; Celli, B.R. Systemic manifestations and comorbidities of COPD. Eur. Respir. J. 2009, 33, 1165–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.J.; Lopez, A.D. Alternative projections of mortality and disability by cause 1990–2020. Global Burden Disease Study. Lancet 1997, 349, 1498–1504. [Google Scholar] [CrossRef]
- Decramer, M.; Rennard, S.; Troosters, T.; Mapel, D.W.; Giardino, N.; Mannino, D.; Wouters, E.; Sethi, S.; Cooper, C.B. COPD as a lung disease with systemic consequences clinical impact, mechanisms, and potential for early intervention. COPD 2008, 5, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Tirlapur, V.G.; Mir, M.A. Nocturnal hypoxemia and associated electrocardiographic changes in patients with chronic obstructive airways disease. N. Engl. J. Med. 1982, 306, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Chen, G.Y.; Kuo, C.D. Hypoxemia and autonomic nervous dysfunction in patients with chronic obstructive pulmonary disease. Respir. Med. 2006, 100, 1547–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, S.K.; De, S. Cardiovascular autonomic neuropathy in chronic obstructive pulmonary disease. Respir. Med. 2005, 99, 126–133. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Top 10 Causes of Death, World Health Organisation. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 10 July 2019).
- Huiart, L.; Ernst, P.; Suissa, S. Cardiovascular morbidity and mortality in COPD. Chest 2005, 128, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Curkendall, S.M.; DeLuise, C.; Jones, J.K.; Lanes, S.; Stang, M.R.; Goehring, E., Jr.; She, D. Cardiovascular disease in patients with chronic obstructive pulmonary disease, Saskatchewan Canada cardiovascular disease in COPD patients. Ann. Epidemiol. 2006, 16, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Claudio, J.S.; Nicola, S.; Claudio, P.; Stefano, N.; Dina, V.; Antonio, S. When kidneys and lungs suffer together. J. Nephrol. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bathoorn, E.; Liesker, J.J.; Postma, D.S.; Boorsma, M.; Bondesson, E.; Koëter, G.H.; Kauffman, H.F.; van Oosterhout, A.J.; Kerstjens, H.A. Anti-inflammatory effects of combined budesonide/formoterol in COPD exacerbations. COPD 2008, 5, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.I.; Barbera, J.A.; Ramirez, J.; Gomez, F.P.; Roca, J.; Jover, L.; Gimferrer, J.M.; Rodriguez-Roisin, R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 274, L908–L913. [Google Scholar] [CrossRef] [PubMed]
- Perera, W.R.; Hurst, J.R.; Wilkinson, T.M.; Sapsford, R.J.; Müllerova, H.; Donaldson, G.C.; Wedzicha, J.A. Inflammatory changes, recovery and recurrence at COPD exacerbation. Eur. Respir. J. 2007, 29, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockley, R.A.; Mannino, D.; Barnes, P.J. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and immune response in COPD: Where do we stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [PubMed]
- Falk, J.A.; Kadiev, S.; Criner, G.J.; Scharf, S.M.; Minai, O.A.; Diaz, P. Cardiac disease in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2008, 5, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, E.; Barbeta, E.; Ielpo, A.; Torres, A. Management of severe acute exacerbations of COPD: An updated narrative review. Multidiscip. Respir. Med. 2018, 13, 36. [Google Scholar] [CrossRef]
- Yohannes, A.M.; Connolly, M.J. Pulmonary rehabilitation programmes in the UK: A national representative survey. Clin. Rehabil. 2004, 18, 444–449. [Google Scholar] [CrossRef]
- Ries, A.L.; Kaplan, R.M.; Myers, R.; Prewitt, L.M. Maintenance after pulmonary rehabilitation in chronic lung disease: A randomized trial. Am. J. Respir. Crit. Care Med. 2003, 167, 880–888. [Google Scholar] [CrossRef]
- Casaburi, R.; Pórszász, J.; Burns, M.R.; Carithers, E.R.; Chang, R.S.; Cooper, C.B. Physiologic benefits of exercise training in rehabilitation of patients with severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1997, 155, 1541–1551. [Google Scholar] [CrossRef]
- Vivodtzev, I.; Minet, C.; Wuyam, B.; Borel, J.-C.; Vottero, G.; Monneret, D.; Baguet, J.-P.; Lévy, P.; Pépin, J.-L. Significant improvement in arterial stiffness after endurance training in patients with COPD. Chest 2010, 137, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.S.; Duckers, J.M.; Enright, S.; Cockcroft, J.R.; Shale, D.J.; Bolton, C.E. Does pulmonary rehabilitation address cardiovascular risk factors in patients with COPD? BMC Pulm. Med. 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.M.; Kuebler, W.M. Endothelial cell regulation of pulmonary vascular tone, inflammation, and coagulation. Compr. Physiol. 2015, 5, 531–559. [Google Scholar] [PubMed]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Laucho-Contreras, M.E.; Petersen, H.; Bijol, V.; Sholl, L.M.; Choi, M.E.; Divo, M.; Pinto-Plata, V.; Chetta, A.; Tesfaigzi, Y.; et al. A Pilot Study Linking Endothelial Injury in Lungs and Kidneys in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, A.; Neveu, D.; Vachier, I.; Paganin, F.; Godard, P.; Chanez, P. Specificity of basement membrane thickening in severe asthma. J. Allergy Clin. Immunol. 2007, 119, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Liesker, J.J.; Ten Hacken, N.H.; Zeinstra-Smith, M.; Rutgers, S.R.; Postma, D.S.; Timens, W. Reticular basement membrane in asthma and COPD: Similar thickness, yet different composition. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 127–135. [Google Scholar] [Green Version]
- Soltani, A.; Reid, D.W.; Sohal, S.S.; Wood-Baker, R.; Weston, S.; Muller, H.K. Walters EH Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: A cross-sectional study. Respir. Res. 2010, 11, 105. [Google Scholar] [CrossRef]
- Soltani, A.; Muller, H.K.; Sohal, S.S.; Reid, D.W.; Weston, S.; Wood-Baker, R.; Walters, E.H. Distinctive characteristics of bronchial reticular basement membrane and vessel remodelling in chronic obstructive pulmonary disease (COPD) and in asthma: They are not the same disease. Histopathology 2012, 60, 964–970. [Google Scholar] [CrossRef]
- Arafah, M.A.; Raddaoui, E.; Kassimi, F.A.; Alhamad, E.H.; Alboukai, A.A.; Alshedoukhy, A.A.; Ouban, A. Endobronchial biopsy in the final diagnosis of chronic obstructive pulmonary disease and asthma: A clinicopathological study. Ann. Saudi Med. 2018, 38, 118–124. [Google Scholar] [CrossRef]
- Kutcher, M.E.; Herman, I.M. The pericyte: Cellular regulator of microvascular blood flow. Microvasc. Res. 2009, 77, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gane, J.; Stockley, R. Mechanisms of neutrophil transmigration across the vascular endothelium in COPD. Thorax 2012, 67, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Vandivier, R.W.; Douglas, I.S. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc. Am. Thorac. Soc. 2006, 3, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Segura-Valdez, L.; Pardo, A.; Gaxiola, M.; Uhal, B.D.; Becerril, C.; Selman, M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000, 117, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Gharib, S.A.; Manicone, A.M.; Parks, W.C. Matrix metalloproteinases in emphysema. Matrix Biol. 2018, 73, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Gilowska, I.; Kasper, Ł.; Bogacz, K.; Szczegielniak, J.; Szymasek, T.; Kasper, M.; Czerwinski, M.; Sładek, K.; Majorczyk, E. Impact of Matrix Metalloproteinase 9 on COPD Development in Polish Patients: Genetic Polymorphism, Protein Level, and Their Relationship with Lung Function. Biomed. Res. Int. 2018, 2018, 6417415. [Google Scholar] [CrossRef] [PubMed]
- Kraen, M.; Frantz, S.; Nihlén, U.; Engström, G.; Löfdahl, C.G.; Wollmer, P.; Dencker, M. Matrix Metalloproteinases in COPD and atherosclerosis with emphasis on the effects of smoking. PLoS ONE 2019, 14, e0211987. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Autophagy, selective autophagy, and necroptosis in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Yokohori, N.; Aoshiba, K.; Nagai, A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Respiratory Failure Research Group in Japan. Chest 2004, 125, 626–632. [Google Scholar] [CrossRef]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Kyomoto, Y.; Kanazawa, H.; Tochino, Y.; Watanabe, T.; Asai, K.; Kawaguchi, T. Possible role of airway microvascular permeability on airway obstruction in patients with chronic obstructive pulmonary disease. Respir. Med. 2019, 146, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Wanner, A.; Mendes, E.S. Airway endothelial dysfunction in asthma and chronic obstructive pulmonary disease: A challenge for future research. Am. J. Respir. Crit. Care Med. 2010, 182, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Kuźnar-Kamińska, B.; Mikuła-Pietrasik, J.; Mały, E.; Makowska, N.; Malec, M.; Tykarski, A.; Batura-Gabryel, H.; Książek, K. Serum from patients with chronic obstructive pulmonary disease promotes proangiogenic behavior of the vascular endothelium. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7470–7481. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartjes, F.J.; Vonk, J.M.; Faiz, A.; Hiemstra, P.S.; Lapperre, T.S.; Kerstjens, H.A.M.; Postma, D.S.; van den Berge, M. Predictive value of eosinophils and neutrophils on clinical effects of ICS in COPD. Respirology 2018, 23, 1023–1031. [Google Scholar] [CrossRef]
- Capron, T.; Bourdin, A.; Perez, T.; Chanez, P. COPD beyond proximal bronchial obstruction: Phenotyping and related tools at the bedside. Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Lüscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef]
- Mocsai, A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J. Exp. Med. 2013, 210, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Klappacher, M.; Kofler, B.; Steinbacher, P.; Vitkov, L.; Grabcanovic-Musija, F.; Studnicka, M. New Aspects on the Structure of Neutrophil Extracellular Traps from Chronic Obstructive Pulmonary Disease and In Vitro Generation. PLoS ONE 2014, 9, e97784. [Google Scholar] [CrossRef] [PubMed]
- Twaddell, S.H.; Baines, K.J.; Grainge, C.; Gibson, P.G. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 2019. [Google Scholar] [CrossRef] [PubMed]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, WD.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.A. Neutrophils and the pathogenesis of COPD. Chest 2002, 121 (Suppl. 5), 151S–155S. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, I.S.; Bayley, D.L.; Lalor, P.; Adams, D.H.; Stockley, R.A. Endothelial interactions of neutrophils under flow in chronic obstructive pulmonary disease. Eur. Respir. J. 2005, 25, 612–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giavazzi, R.; Nicoletti, M.I.; Chirivi, R.G.; Hemingway, I.; Bernasconi, S.; Allavena, P.; Gearing, A.J. Soluble intercellular adhesion molecule-1 (ICAM-1) is released into the serum and ascites of human ovarian carcinoma patients and in nude mice bearing tumour xenografts. Eur. J. Cancer 1994, 30, 1865–1870. [Google Scholar] [CrossRef]
- Aaron, C.P.; Schwartz, J.E.; Bielinski, S.J.; Hoffman, E.A.; Austin, J.H.; Oelsner, E.C.; Donohue, K.M.; Kalhan, R.; Berardi, C.; Kaufman, J.D.; et al. Intercellular adhesion molecule 1 and progression of percent emphysema: The MESA Lung Study. Respir. Med. 2015, 109, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Hansbro, P.M.; Walters, E.H. Blocking rhinoviral adhesion molecule (ICAM-1): Potential to prevent COPD exacerbations. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1413–1414. [Google Scholar] [CrossRef] [PubMed]
- Oelsner, E.C.; Pottinger, T.D.; Burkart, K.M.; Allison, M.; Buxbaum, S.G.; Hansel, N.N.; Kumar, R.; Larkin, E.K.; Lange, L.A.; Loehr, L.R.; et al. Adhesion molecules, endothelin-1 and lung function in seven population-based cohorts. Biomarkers 2013, 18, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K.; et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Starnes, T.W.; Huttenlocher, A. Neutrophil reverse migration becomes transparent with zebrafish. Adv. Hematol. 2012, 2012, 398640. [Google Scholar] [CrossRef]
- Nourshargh, S.; Renshaw, S.A.; Imhof, B.A. Reverse Migration of Neutrophils: Where, When, How, and Why? Trends Immunol. 2016, 37, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bassler, K.; Aschenbrenner, A.C.; Fujii, W.; Schultze, J.L. Dysregulated Functions of Lung Macrophage Populations in COPD. J. Immunol. Res. 2018, 2018, 2349045. [Google Scholar] [CrossRef]
- Bazzan, E.; Turato, G.; Tinè, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Xie, L.; Lu, J.; Sun, S. Characteristics and potential role of M2 macrophages in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 3029–3039. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Bafadhel, M.; Pavord, I.D.; Russell, R.E.K. Eosinophils in COPD: Just another biomarker? Lancet Respir. Med. 2017, 5, 747–759. [Google Scholar] [CrossRef]
- Kolsum, U.; Donaldson, G.C.; Singh, R.; Barker, B.L.; Gupta, V.; George, L.; Webb, A.J.; Thurston, S.; Brookes, A.J.; McHugh, T.D.; et al. Blood and sputum eosinophils in COPD; relationship with bacterial load. Respir. Res. 2017, 18, 88. [Google Scholar] [CrossRef] [PubMed]
- Kolsum, U.; Southworth, T.; Jackson, N.; Singh, D. Blood eosinophil counts in COPD patients compared to controls. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Fuschillo, S.; Molino, A.; Stellato, C.; Motta, A.; Maniscalco, M. Blood eosinophils as biomarkers of therapeutic response to chronic obstructive pulmonary disease: Still work in progress. Eur. J. Intern. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Kharitonov, S.A.; Ciabattoni, G.; Barnes, P.J. Exhaled leukotrienes and prostaglandins in COPD. Thorax 2003, 58, 585–588. [Google Scholar] [CrossRef] [Green Version]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef]
- Navratilova, Z.; Kolek, V.; Petrek, M. Matrix Metalloproteinases and Their Inhibitors in Chronic Obstructive Pulmonary Disease. Arch. Immunol. Ther. Exp. (Warsz) 2016, 64, 177–193. [Google Scholar] [CrossRef]
- Dahl, M.; Tybjaerg-Hansen, A.; Vestbo, J.; Lange, P.; Nordestgaard, B.G. Elevated plasma fibrinogen associated with reduced pulmonary function and increased risk of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1008–1011. [Google Scholar] [CrossRef]
- Dahl, M.; Vestbo, J.; Lange, P.; Bojesen, S.E.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. C-reactive protein as a predictor of prognosis in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 250–255. [Google Scholar] [CrossRef]
- Su, B.; Liu, T.; Fan, H.; Chen, F.; Ding, H.; Wu, Z.; Wang, H.; Hou, S. Inflammatory Markers and the Risk of Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0150586. [Google Scholar] [CrossRef]
- Cheng, D.T.; Kim, D.K.; Cockayne, D.A.; Belousov, A.; Bitter, H.; Cho, M.H.; Duvoix, A.; Edwards, L.D.; Lomas, D.A.; Miller, B.E.; et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 188, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Atochina-Vasserman, E.N.; Holz, O.; Beers, M.F.; Erpenbeck, V.J.; Krug, N.; Roepcke, S.; Lauer, G.; Elmlinger, M.; Hohlfeld, J.M. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir. Res. 2011, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Coxson, H.O.; Dirksen, A.; Edwards, L.D.; Yates, J.C.; Agusti, A.; Bakke, P.; Calverley, P.M.; Celli, B.; Crim, C.; Duvoix, A.; et al. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: A prospective analysis from the ECLIPSE study. Lancet Respir. Med. 2013, 1, 129–136. [Google Scholar] [CrossRef]
- Mackay, C.R. Chemokines: immunology’s high impact factors. Nat. Immunol. 2001, 2, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Youn, B.S.; Mantel, C.; Broxmeyer, H.E. Chemokines, chemokine receptors and hematopoiesis. Immunol. Rev. 2000, 177, 150–174. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Arenberg, D.A.; Addison, C.L.; Ehlert, J.E.; Burdick, M.D.; Strieter, R.M. CXC chemokines in angiogenesis. J. Leukoc. Biol. 2000, 68, 1–8. [Google Scholar]
- Bradford, E.; Jacobson, S.; Varasteh, J.; Comellas, A.P.; Woodruff, P.; O’Neal, W.; DeMeo, D.L.; Li, X.; Kim, V.; Cho, M.; et al. The value of blood cytokines and chemokines in assessing COPD. Respir. Res. 2017, 18, 180. [Google Scholar] [CrossRef]
- Barnes, P.J. The Cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Bianco, A.; Mazzarella, G.; Turchiarelli, V.; Nigro, E.; Corbi, G.; Scudiero, O.; Sofia, M.; Daniele, A. Adiponectin: An attractive marker for metabolic disorders in Chronic Obstructive Pulmonary Disease (COPD). Nutrients 2013, 5, 4115–4125. [Google Scholar] [CrossRef]
- Li, D.; Wu, Y.; Tian, P.; Zhang, X.; Wang, H.; Wang, T.; Ying, B.; Wang, L.; Shen, Y.; Wen, F. Adipokine CTRP-5 as a Potential Novel Inflammatory Biomarker in Chronic Obstructive Pulmonary Disease. Medicine (Baltimore) 2015, 94, e1503. [Google Scholar] [CrossRef]
- Yoon, H.I.; Li, Y.; Man, S.F.; Tashkin, D.; Wise, R.A.; Connett, J.E.; Anthonisen, N.A.; Churg, A.; Wright, J.L.; Sin, D.D. The complex relationship of serum adiponectin to COPD outcomes COPD and adiponectin. Chest 2012, 142, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.M.; Jeong, B.H.; Woo, S.Y.; Kim, S.Y.; Kim, H.; Lee, J.H.; Lim, S.Y.; Rhee, C.K.; Yoo, K.H.; Lee, J.H.; et al. KOLD Study Group. Association of plasma adipokines with chronic obstructive pulmonary disease severity and progression. Ann. Am. Thorac. Soc. 2015, 12, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Hayashikawa, Y.; Iwata, M.; Inomata, M.; Kawagishi, Y.; Tokui, K.; Taka, C.; Kambara, K.; Okazawa, S.; Yamada, T.; Hayashi, R.; et al. Association of serum adiponectin with asthma and pulmonary function in the Japanese population. Endocr. J. 2015, 62, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.L.; Li, F.; Liu, Y.W.; Shi, Y.J.; Li, Z.H.; Cao, G.K.; Zhu, W. Adiponectin attenuates endoplasmic reticulum stress and alveolar epithelial apoptosis in COPD rats. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4999–5007. [Google Scholar] [PubMed]
- Kamiide, Y.; Inomata, N.; Furuya, M.; Yada, T. Ghrelin ameliorates catabolic conditions and respiratory dysfunction in a chronic obstructive pulmonary disease model of chronic cigarette smoke-exposed rats. Eur. J. Pharmacol. 2015, 755, 88–94. [Google Scholar] [CrossRef]
- Henrot, P.; Prevel, R.; Berger, P.; Dupin, I. Chemokines in COPD: From Implication to Therapeutic Use. Int. J. Mol. Sci. 2019, 20, 2785. [Google Scholar] [CrossRef]
- Galzi, J.-L.; Hachet-Haas, M.; Bonnet, D.; Daubeuf, F.; Lecat, S.; Hibert, M.; Haiech, J.; Frossard, N. Neutralizing endogenous chemokines with small molecules. Principles and potential therapeutic applications. Pharmacol. Ther. 2010, 126, 39–55. [Google Scholar] [CrossRef]
- Proudfoot, A.E.I. Chemokines and Glycosaminoglycans. Front. Immunol. 2015, 6, 246. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, eaah6650. [Google Scholar] [CrossRef]
- Liebow, A.A. Pulmonary emphysema with special reference to vascular changes. Am. Rev. Respir. Dis. 1959, 80, 67–93. [Google Scholar] [PubMed]
- Voelkel, N.F.; Gomez-Arroyo, J. Mizuno S COPD/emphysema: The vascular story. Pulm. Circ. 2011, 1, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163 Pt 1, 737–744. [Google Scholar] [CrossRef]
- Dinh-Xuan, A.T.; Higenbottam, T.W.; Clelland, C.A.; Pepke-Zaba, J.; Cremona, G.; Butt, A.Y.; Large, S.R.; Wells, F.C.; Wallwork, J. Impairment of endothelium-dependent pulmonary-artery Pulmonary Circulation Volume 8 Number 1 | 13 relaxation in chronic obstructive lung disease. N. Engl. J. Med. 1991, 324, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Washko, G.R. The role and potential of imaging in COPD. Med. Clin. 2012, 96, 729–743. [Google Scholar] [CrossRef]
- Peinado, V.I.; Pizarro, S.; Barberà, J.A. Pulmonary vascular involvement in COPD. Chest 2008, 134, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yoshikawa, J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest 2005, 128, 3191–3197. [Google Scholar] [CrossRef]
- Noe, J.; Petrusca, D.; Rush, N.; Deng, P.; VanDemark, M.; Berdyshev, E.; Gu, Y.; Smith, P.; Schweitzer, K.; Pilewsky, J.; et al. CFTR regulation of intracellular pH and ceramides is required for lung endothelial cell apoptosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 314–323. [Google Scholar] [CrossRef]
- Brown, M.B.; Hunt, W.R.; Noe, J.E.; Rush, N.I.; Schweitzer, K.S.; Leece, T.C.; Petrache, I. Loss of Cystic Fibrosis Transmembrane Conductance Regulator Impairs Lung Endothelial Cell Barrier Function and Increases Susceptibility to Microvascular Damage from Cigarette Smoke. Pulm. Circ. 2014, 4, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1- antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2016, 17, 1801. [Google Scholar] [CrossRef] [PubMed]
- Lockett, A.D.; Brown, M.B.; Santos-Falcon, N.; Rush, N.I.; Oueini, H.; Oberle, A.J.; Bolanis, E.; Fragoso, M.A.; Petrusca, D.N.; Serban, K.A.; et al. Active trafficking of alpha 1 antitrypsin across the lung endothelium. PLoS ONE 2014, 9, e93979. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, S.; Ren, H.; Sun, W.; Zhang, H.; Sun, J.; Yang, S.; Hao, J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014, 74, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp. Ther. Med. 2018, 16, 4553–4561. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, A.A.; Aragonés, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6, E68. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290. [Google Scholar] [CrossRef]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Cruden, N.L.; Amer, D.A.; Li, V.K.; Goudie, E.B.; Johnston, N.R.; Sharma, S.; Neilson, I.; Webb, D.J.; Megson, I.L.; et al. Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 2008, 52, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Lee, S.H.; Kim, C.H.; Yang, K.S.; Lee, E.J.; Min, K.H.; Hur, G.Y.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; et al. Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1alpha in lung tissue of patients with chronic bronchitis. Clin. Biochem. 2014, 47, 552–559. [Google Scholar] [CrossRef]
- McCarty, G.; Awad, O.; Loeb, D.M. WT1 protein directly regulates expression of vascular endothelial growth factor and is a mediator of tumor response to hypoxia. J. Biol. Chem. 2011, 286, 43634–43643. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, A.R.; de Boer, W.I.; Alagappan, V.K.; Sterk, P.J.; Sharma, H.S. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax 2005, 60, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, S.; Peinado, V.I.; Ramirez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1250–1256. [Google Scholar] [CrossRef]
- Campisi, J.; Fagagna, F.D.A. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Amsellem, V.; Gary-Bobo, G.; Marcos, E.; Maitre, B.; Chaar, V.; Validire, P.; Stern, J.B.; Noureddine, H.; Sapin, E.; Rideau, D.; et al. Obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1358–1366. [Google Scholar] [CrossRef]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Zakynthinos, E.; Daniil, Z.; Papanikolaou, J.; Makris, D. Pulmonary hypertension in COPD: Pathophysiology and therapeutic targets. Curr. Drug Targets 2011, 12, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, B.G.; Bogaard, J.H.; Trip, P.; Groepenhoff, H.; Rietema, H.; Holverda, S.; Boonstra, A.; Postmus, P.E.; Westerhof, N.; Vonk-Noordegraaf, A. Ventilatory and cardiocirculatory exercise profiles in COPD: The role of pulmonary hypertension. Chest 2012, 142, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.; Faller, M.; Fourgaut, G.; Mennecier, B.; Weitzenblum, E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 159, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Thabut, G.; Dauriat, G.; Stern, J.B.; Logeart, D.; Lévy, A.; Marrash-Chahla, R.; Mal, H. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest 2005, 127, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Weir-McCall, J.R.; Struthers, A.D.; Lipworth, B.J.; Houston, J.G. The role of pulmonary arterial stiffness in COPD. Respir. Med. 2015, 109, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Weitzenblum, E. Chronic cor pulmonale. Heart 2003, 89, 225–230. [Google Scholar] [CrossRef]
- Barbera, J.A. Mechanisms of development of chronic obstructive pulmonary disease-associated pulmonary hypertension. Pulm. Circ. 2013, 3, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.C.; Keane, M.P.; Gaine, S.; McLoughlin, P. Hypoxic pulmonary hypertension in chronic lung diseases: Novel vasoconstrictor pathways. Lancet Respir. Med. 2016, 4, 225–236. [Google Scholar] [CrossRef]
- Borgas, D.; Chambers, E.; Newton, J.; Ko, J.; Rivera, S.; Rounds, S.; Lu, Q. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am. J. Respir. Cell Mol. Biol. 2016, 54, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Maclay, J.D.; McAllister, D.A.; Mills, N.L.; Paterson, F.P.; Ludlam, C.A.; Drost, E.M.; Newby, D.E.; Macnee, W. Vascular dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 180, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Paganin, F.; Capieu, L.; Scichilone, N.; Bellia, M.; Maakel, L.; Bellia, V.; Godard, P.; Bousquet, J.; Chanez, P. Airway remodelling assessed by sputum and high-resolution computed tomography in asthma and COPD. Eur. Respir. J. 2004, 24, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Brightling, C.E.; Monteiro, W.; Ward, R.; Parker, D.; Morgan, M.D.; Wardlaw, A.J.; Pavord, I.D. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: A randomised controlled trial. Lancet 2000, 356, 1480–1485. [Google Scholar] [CrossRef]
- Pizzichini, E.; Pizzichini, M.M.; Gibson, P.; Parameswaran, K.; Gleich, G.J.; Berman, L.; Dolovich, J.; Hargreave, F.E. Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am. J. Respir. Crit. Care Med. 1998, 158, 1511–1517. [Google Scholar] [CrossRef]
- Sakao, S.; Voelkel, N.F.; Tatsumi, K. The vascular bed in COPD: Pulmonary hypertension and pulmonary vascular alterations. Eur. Respir. Rev. 2014, 23, 350–355. [Google Scholar] [CrossRef]
- Biondi-Zoccai, G.; Sciarretta, S.; Bullen, C.; Nocella, C.; Violi, F.; Loffredo, L.; Pignatelli, P.; Perri, L.; Peruzzi, M.; Marullo, A.G.M.; et al. Acute Effects of Heat-Not-Burn, Electronic Vaping, and Traditional Tobacco Combustion Cigarettes: The Sapienza University of Rome-Vascular Assessment of Proatherosclerotic Effects of Smoking (SUR-VAPES) 2 Randomized Trial. J. Am. Heart Assoc. 2019, 8, e010455. [Google Scholar] [CrossRef]
- Gonzalez, S.; Hards, J.; van Eeden, S.; Hogg, J.C. The expression of adhesion molecules in cigarette smoke-induced airways obstruction. Eur. Respir. J. 1996, 9, 1995–2001. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.F.; Hwang, S.-J.; Vasan, R.S.; Larson, M.G.; Pencina, M.J.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J. Arterial stiffness and cardiovascular events: The Framingham Heart Study. Circulation 2010, 121, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.S.; Briet, M. Arterial Stiffness and Chronic Kidney Disease. Pulse (Basel) 2016, 3, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursula Quinn, U.; Tomlinson, L.A.; Cockcroft, J.R. Arterial stiffness. JRSM Cardiovasc. Dis. 2012, 1, Cvd.2012.012024. [Google Scholar]
- Smith, M.C.; Wrobel, J.P. Epidemiology and clinical impact of major comorbidities in patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2014, 9, 871–888. [Google Scholar] [CrossRef] [PubMed]
- Coulson, J.M.; Rudd, J.H.; Duckers, J.M.; Rees, J.I.; Shale, D.J.; Bolton, C.E.; Cockcroft, J.R. Excessive aortic inflammation in chronic obstructive pulmonary disease: An 18F-FDG PET pilot study. J. Nucl. Med. 2010, 51, 1357–1360. [Google Scholar] [CrossRef]
- Szűcs, B.; Petrekanits, M.; Varga, J. Effectiveness of a 4-week rehabilitation program on endothelial function, blood vessel elasticity in patients with chronic obstructive pulmonary disease. J. Thorac. D 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.E.; Gale, N.S.; Cockcroft, J.R. Exercise and cardiovascular benefit in subjects with COPD: The need for randomised trials. Eur. Respir. J. 2014, 44, 263–264. [Google Scholar] [CrossRef]
- Canavan, J.L.; Kaliaraju, D.; Nolan, C.M.; Clark, A.L.; Jones, S.E.; Kon, S.S.; Polkey, M.I.; Man, W.D. Does pulmonary rehabilitation reduce peripheral blood pressure in patients with chronic obstructive pulmonary disease? Chronic Respir. Dis. 2015, 12, 256–263. [Google Scholar] [CrossRef]
- Vanfleteren, L.E.; Spruit, M.A.; Groenen, M.T.; Bruijnzeel, P.L.; Taib, Z.; Rutten, E.P.; Op ’t Roodt, J.; Akkermans, M.A.; Wouters, E.F.; Franssen, F.M. Arterial stiffness in patients with COPD: The role of systemic inflammation and the effects of pulmonary rehabilitation. Eur. Respir. J. 2014, 43, 1306–1315. [Google Scholar] [CrossRef]

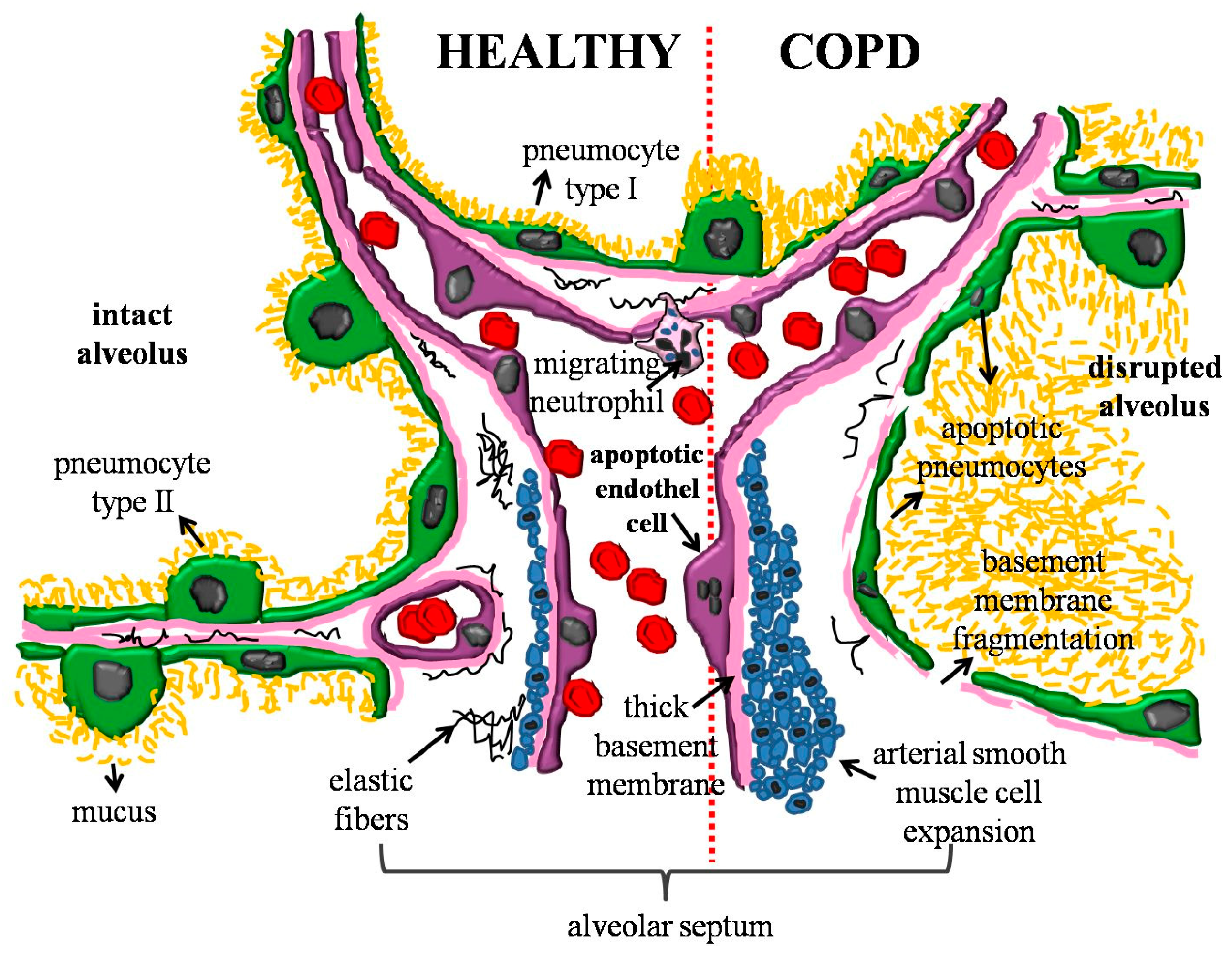



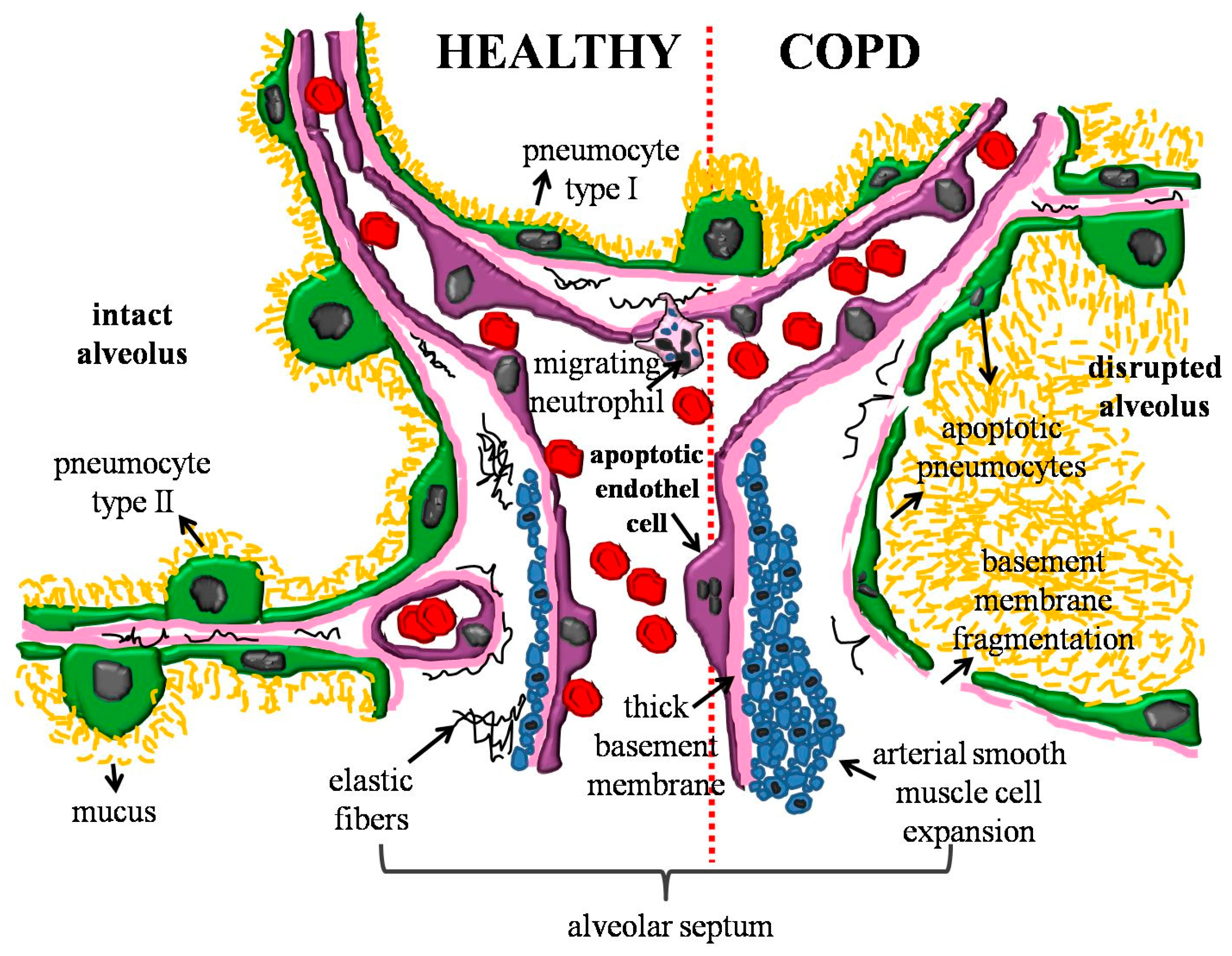
{kind=link}
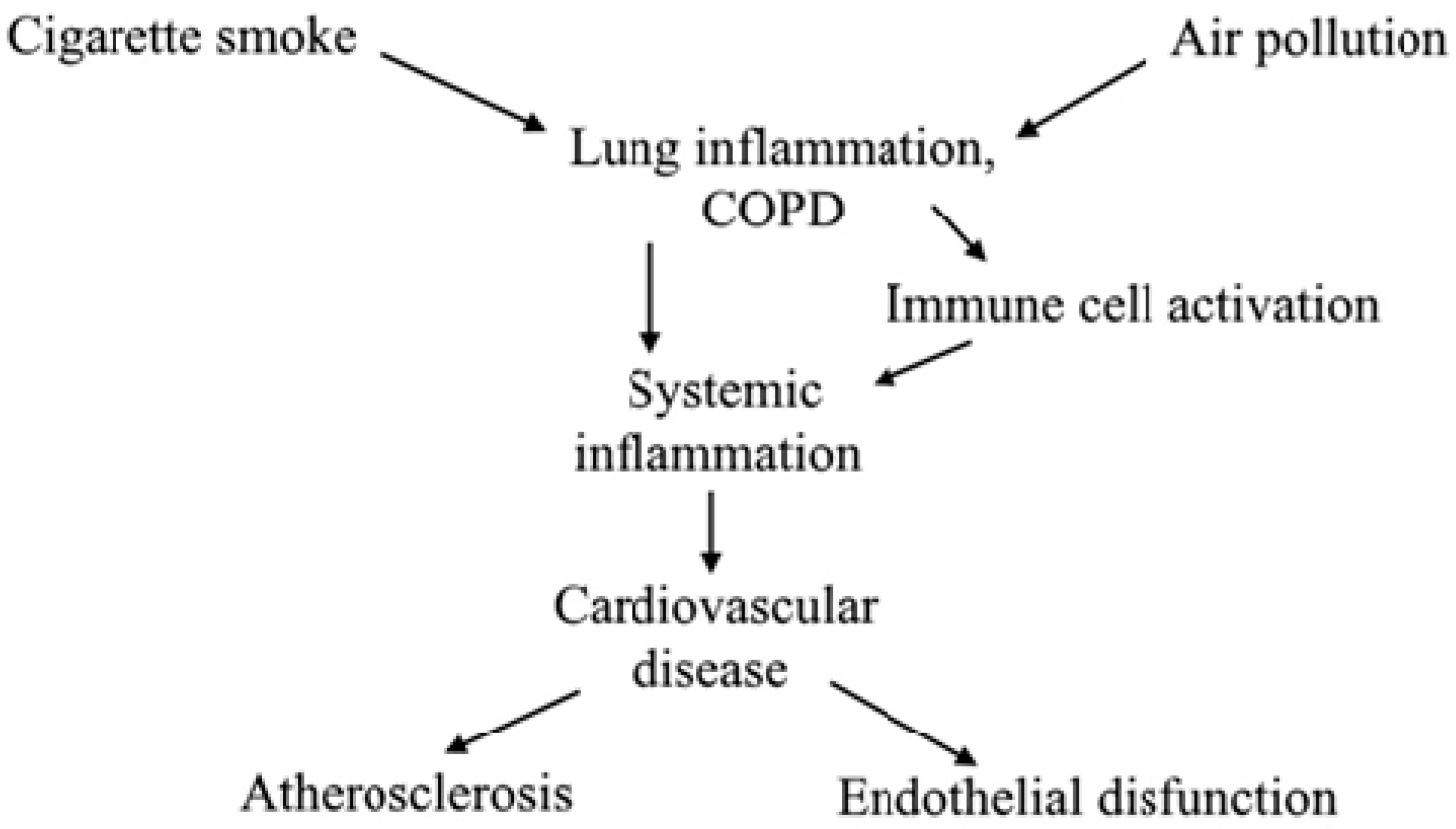
{kind=link}
{kind=link}
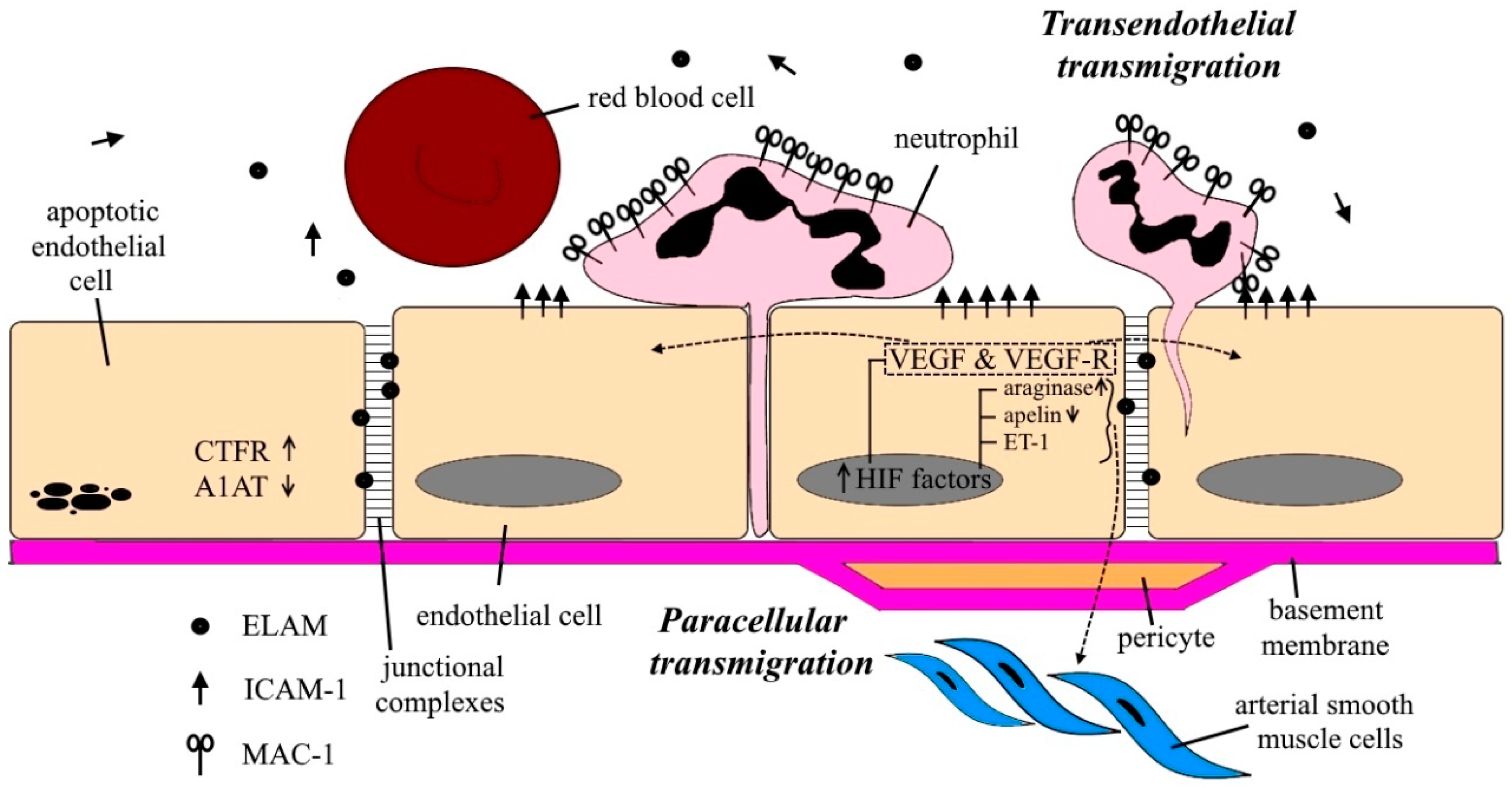{kind=link}
| Parameter | Before Treatment | After Treatment |
|---|---|---|
| Sys (Hgmm) | 133.38 ± 22.15 | 126.48 ± 20.22 |
| Dias (Hgmm) | 76.95 ± 14.37 | 75.4 ±12.7 |
| Pulse (bpm) | 76.95 ± 14.37 | 72.53 ± 13.65 |
| AIX (%) | 3.54 ± 35.59 | 2.93 ± 30.79 |
| PWV (m/s) | 11.74 ± 2.13 | 11.4 ± 2.73 |
| DAI (%) | 46.32 ± 6.81 | 47.1 ± 70.2 |
| FEV1 (l) | 45.43 ± 20.2 | 45.06 ± 18.2 |
| FVC (l) | 75.81 ± 22.71 | 74.78 ± 17.37 |
| mMRC | 1.86 ± 0.71 | 1.63 ± 0.6 * |
| MIP (cmH2O) | 57.72 ± 22.69 | 63.63 ± 18.01 * |
| CWE (cm) | 2.84 ± 1.26 | 4.00 ± 1.76 * |
| BHT (s) | 25.77 ± 10.63 | 29.21 ± 11.60 * |
| GS (kg) | 24.87 ± 11.88 | 27.03 ± 11.43 * |
| 6MWD (m) | 335.32 ± 110.43 | 398.32 ± 126.21 * |
| CAT | 17.00 ± 8.49 | 11.89 ± 7.31 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. Int. J. Mol. Sci. 2019, 20, 4329. https://doi.org/10.3390/ijms20184329
Szucs B, Szucs C, Petrekanits M, Varga JT. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. International Journal of Molecular Sciences. 2019; 20(18):4329. https://doi.org/10.3390/ijms20184329
Chicago/Turabian StyleSzucs, Botond, Csilla Szucs, Mate Petrekanits, and Janos T. Varga. 2019. "Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article" International Journal of Molecular Sciences 20, no. 18: 4329. https://doi.org/10.3390/ijms20184329
APA StyleSzucs, B., Szucs, C., Petrekanits, M., & Varga, J. T. (2019). Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. International Journal of Molecular Sciences, 20(18), 4329. https://doi.org/10.3390/ijms20184329