Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
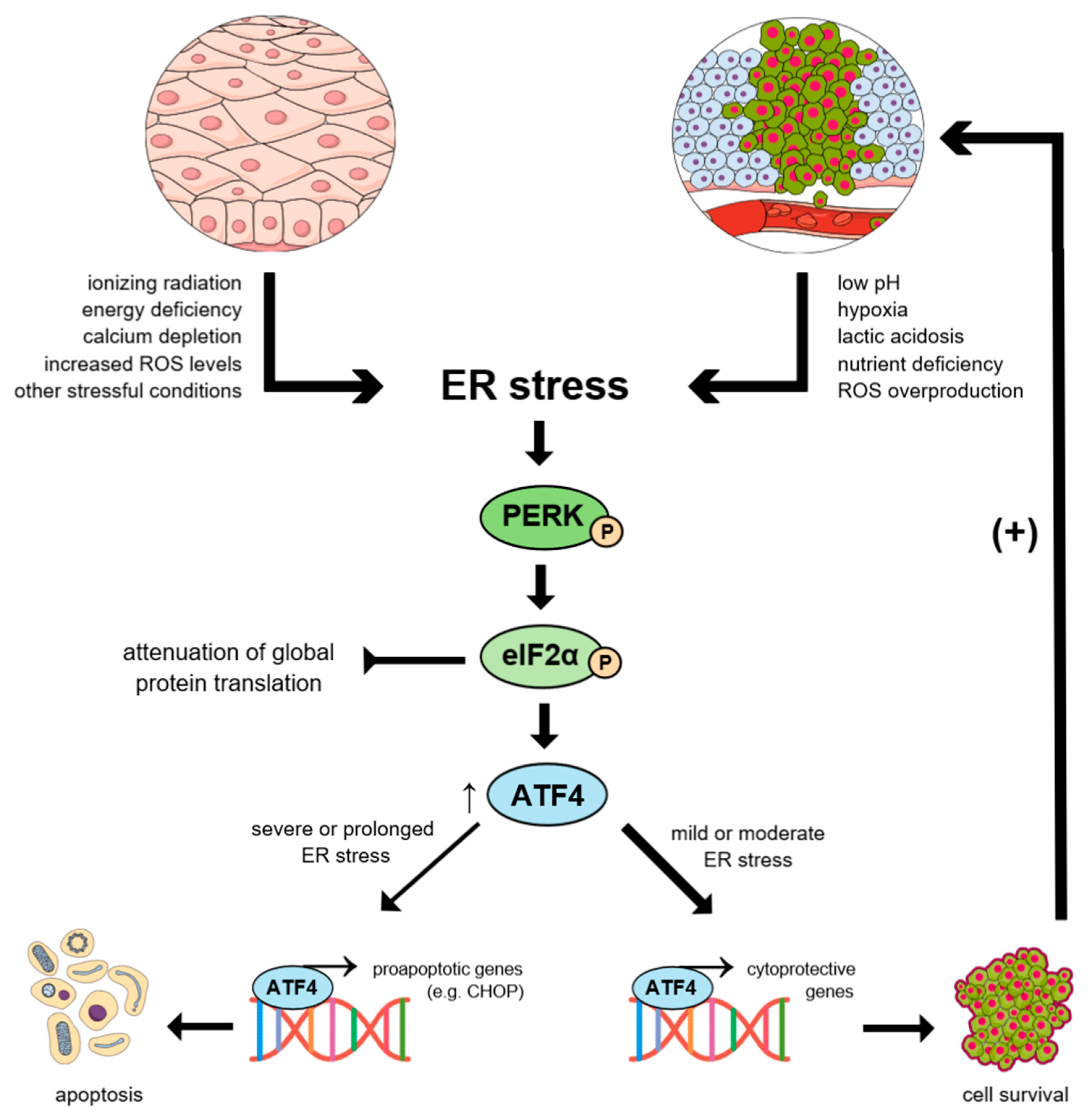
:1. Introduction
2. UPR in Cancer Initiation and Progression
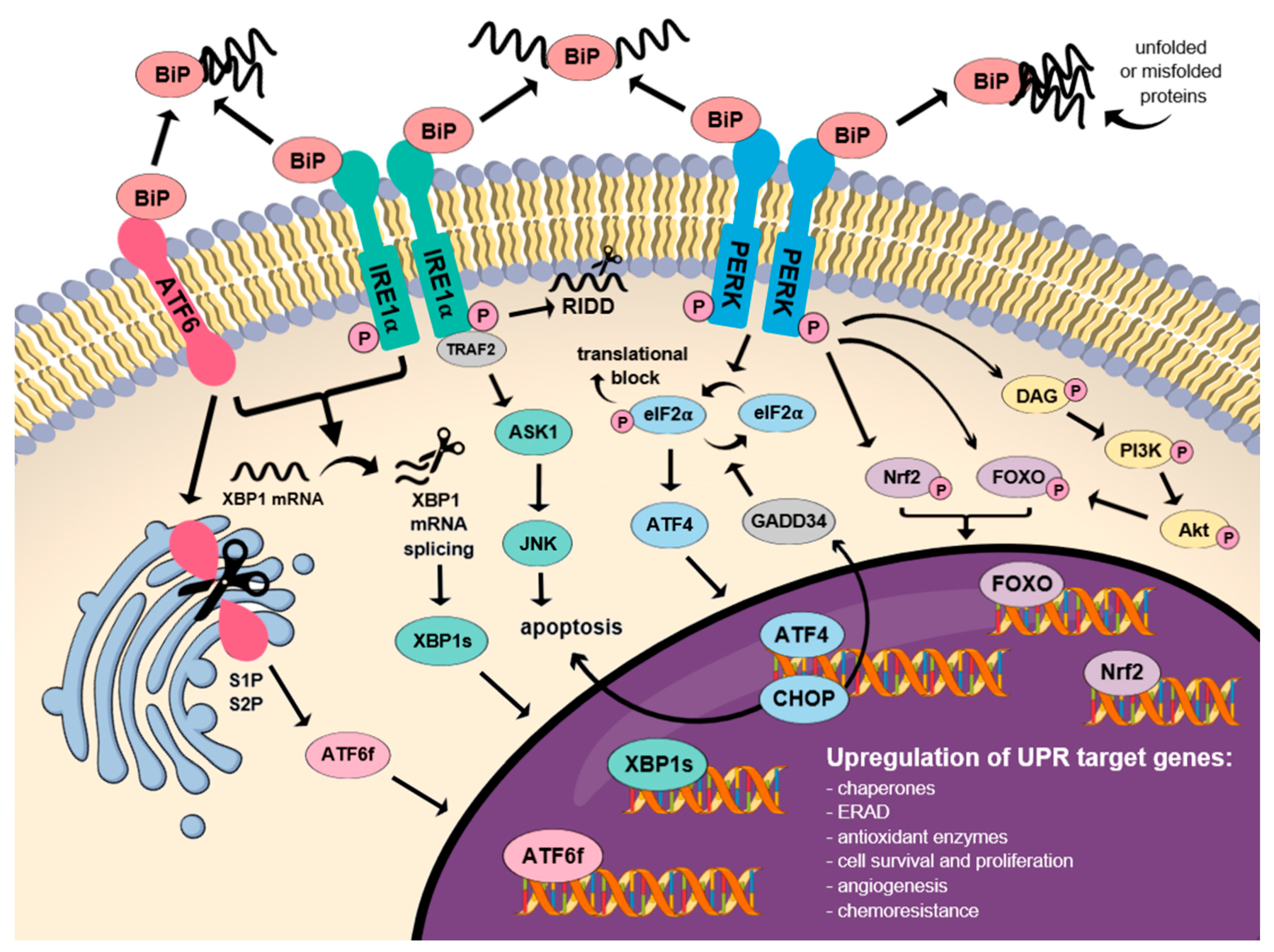
3. Molecular Mechanism of the UPR Signaling Pathway Activation
3.1. BiPs as the Major Regulators of the UPR Signaling Pathway
3.2. Dual Function of the IRE1α Receptor in Cell Fate Determination
3.3. The Role of the ATF6 in Proteostasis Restoration
3.4. Molecular Mechanism of the PERK-mediated Activation of the UPR Signaling Pathway
3.5. Nrf2 is Engaged in Multiple Antioxidant Responses
3.6. Multidimensional Role of FOXOs in Cell Fate
3.7. CHOP Protein and JNK as the Key Factors in Apoptosis Induction
4. Molecular Basis of UPR in Cancer Development
4.1. ER Stress-mediated Activation of the UPR Signaling Pathway in Cancer Cells
4.2. BiPs as the Markers of the UPR Signaling Pathway Activation in Cancer
4.3. IRE1α/XBP1-dependent Regulation of Tumor Growth
4.4. Cytoprotective and Pro-tumorigenic Role of ATF6
4.5. Paradoxical Outcomes of PERK Activation within Cancer Cells
4.6. The Role of ATF4 in Cancer Initiation and Progression
4.7. A critical Role of FOXO Proteins in Carcinogenesis
4.8. Multiple Functions of Apoptotic Proteins in Deciding Cancer Cell Fate
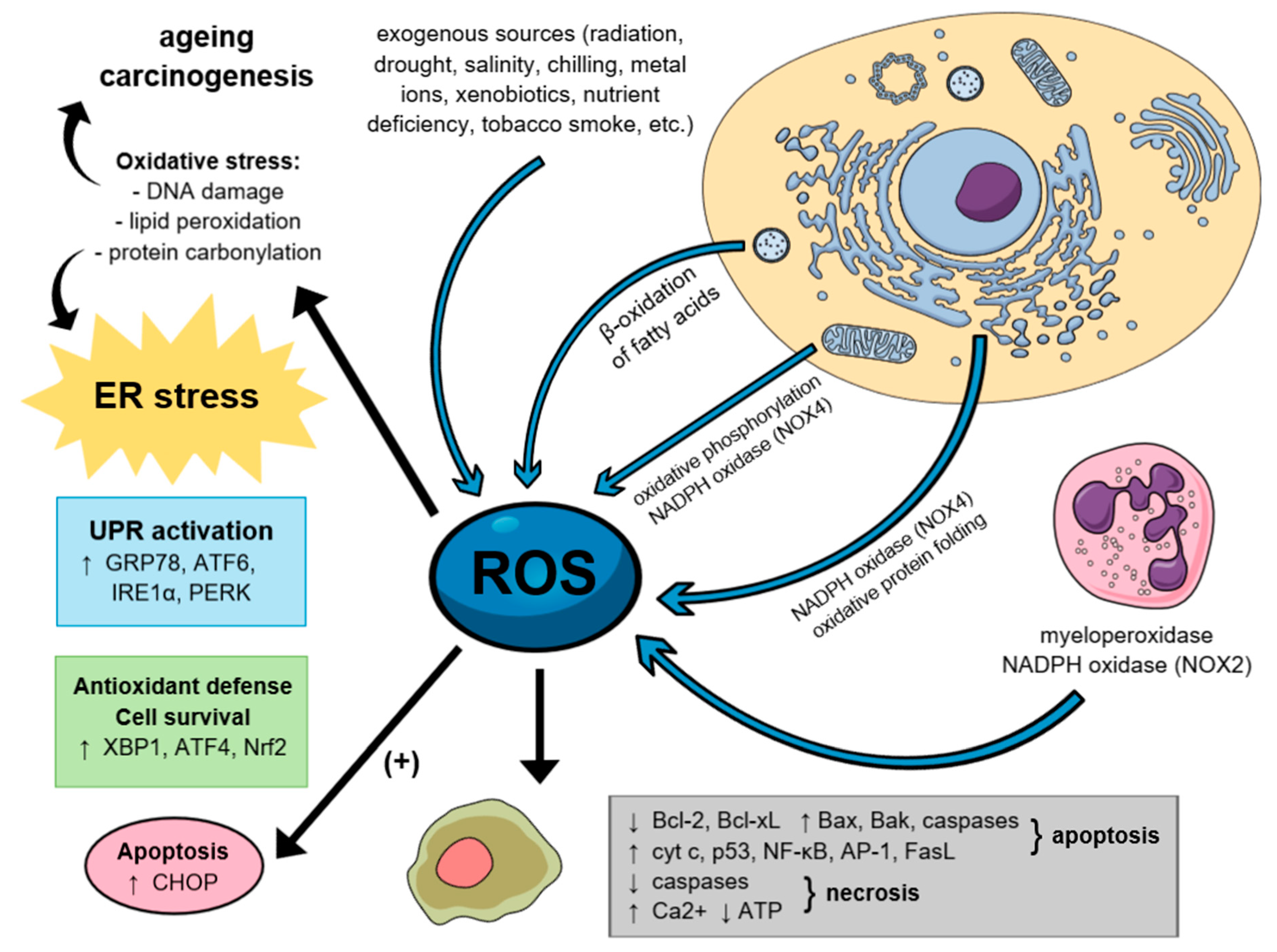
5. The Role of Reactive Oxygen Species in Carcinogenesis
6. Innovative Drugs Depending on Oxidative Mechanisms
7. Summary and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Nowak-Zdunczyk, A.; Lewko, D.; Wojtczak, R.; Diehl, J.A.; Majsterek, I. Breaking the DNA Damage Response via Serine/Threonine Kinase Inhibitors to Improve Cancer Treatment. Curr. Med. Chem. 2019, 26, 1425–1445. [Google Scholar] [CrossRef]
- Kye, B.H.; Cho, H.M. Overview of radiation therapy for treating rectal cancer. Ann. Coloproctol. 2014, 30, 165–174. [Google Scholar] [CrossRef]
- Raman, V.; Jawitz, O.K.; Voigt, S.L.; Farrow, N.E.; Yang, C.J.; D’Amico, T.A.; Harpole, D.H., Jr. Surgery is Associated with Survival Benefit in T4a Esophageal Adenocarcinoma: A National Analysis. Ann. Thorac. Surg. 2019. [Google Scholar] [CrossRef]
- Autore, F.; Sora, F.; Chiusolo, P.; Annunziata, M.; Iurlo, A.; Cattaneo, D.; Galimberti, S.; Bajer, J.A.; Sica, S. ‘Secondary chronic myeloid leukemia’: Comparison between patients previously exposed or not to chemo- and/or radiotherapy. Leuk. Lymphoma 2019, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yoo, R.N.; Kim, H.J. Total neoadjuvant therapy in locally advanced rectal cancer: Role of systemic chemotherapy. Ann. Gastroenterol. Surg. 2019, 3, 356–367. [Google Scholar] [CrossRef]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER Stress In Vivo and In Vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef]
- Naz, S.; Shamoon, M.; Wang, R.; Zhang, L.; Zhou, J.; Chen, J. Advances in Therapeutic Implications of Inorganic Drug Delivery Nano-Platforms for Cancer. Int. J. Mol. Sci. 2019, 20, 965. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors with Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef]
- Qi, L.; Luo, Q.; Zhang, Y.; Jia, F.; Zhao, Y.; Wang, F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef]
- Meirow, D.; Nugent, D. The effects of radiotherapy and chemotherapy on female reproduction. Hum. Reprod. Update 2001, 7, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.M.; Goldberg, R.M. Treatment of metastatic colorectal cancer. Semin. Oncol. 2011, 38, 552–560. [Google Scholar] [CrossRef]
- Zou, H.; Li, L.; Garcia Carcedo, I.; Xu, Z.P.; Monteiro, M.; Gu, W. Synergistic inhibition of colon cancer cell growth with nanoemulsion-loaded paclitaxel and PI3K/mTOR dual inhibitor BEZ235 through apoptosis. Int. J. Nanomed. 2016, 11, 1947–1958. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Hatoum, H.; Dy, G.K. First line treatment of advanced non-small-cell lung cancer—Specific focus on albumin bound paclitaxel. Int. J. Nanomed. 2014, 9, 209–221. [Google Scholar] [CrossRef]
- Bhat, N.; Kalthur, S.G.; Padmashali, S.; Monappa, V. Toxic Effects of Different Doses of Cyclophosphamide on Liver and Kidney Tissue in Swiss Albino Mice: A Histopathological Study. Ethiop. J. Health Sci. 2018, 28, 711–716. [Google Scholar] [CrossRef]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef]
- Bresciani, L.; Orlandi, E.; Piazza, C. Radiation-induced papillary thyroid cancer: Is it a distinct clinical entity? Curr. Opin. Otolaryngol. Head Neck Surg. 2019, 27, 117–122. [Google Scholar] [CrossRef]
- Choi, J.N.; Kim, D.; Choi, H.K.; Yoo, K.M.; Kim, J.; Lee, C.H. 2’-hydroxylation of genistein enhanced antioxidant and antiproliferative activities in mcf-7 human breast cancer cells. J. Microbiol. Biotechnol. 2009, 19, 1348–1354. [Google Scholar] [CrossRef]
- Zhu, Q.; Meisinger, J.; Van Thiel, D.H.; Zhang, Y.; Mobarhan, S. Effects of soybean extract on morphology and survival of Caco-2, SW620, and HT-29 cells. Nutr. Cancer 2002, 42, 131–140. [Google Scholar] [CrossRef]
- Gao, S.; Hu, M. Bioavailability challenges associated with development of anti-cancer phenolics. Mini Rev. Med. Chem. 2010, 10, 550–567. [Google Scholar] [CrossRef]
- Pinho, E.; Grootveld, M.; Soares, G.; Henriques, M. Cyclodextrins as encapsulation agents for plant bioactive compounds. Carbohydr. Polym. 2014, 101, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta 2014, 1843, 2143–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef]
- Rozpedek, W.; Nowak, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. Molecular Basis of Human Diseases and Targeted Therapy Based on Small-Molecule Inhibitors of ER Stress-Induced Signaling Pathways. Curr. Mol. Med. 2017, 17, 118–132. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013, 33, 4683–4694. [Google Scholar]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Amann, T.; Hellerbrand, C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 2009, 13, 1411–1427. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [Green Version]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Mohan, S.; Brown, L.; Ayyappan, P. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Cojocari, D.; Vellanki, R.N.; Sit, B.; Uehling, D.; Koritzinsky, M.; Wouters, B.G. New small molecule inhibitors of UPR activation demonstrate that PERK, but not IRE1α signaling is essential for promoting adaptation and survival to hypoxia. Radiother. Oncol. 2013, 108, 541–547. [Google Scholar] [CrossRef]
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef]
- Oida, T.; Weiner, H.L. Overexpression of TGF-ss 1 gene induces cell surface localized glucose-regulated protein 78-associated latency-associated peptide/TGF-ss. J. Immunol. 2010, 185, 3529–3535. [Google Scholar] [CrossRef]
- Gray, P.C.; Vale, W. Cripto/GRP78 modulation of the TGF-β pathway in development and oncogenesis. FEBS Lett. 2012, 586, 1836–1845. [Google Scholar] [CrossRef]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Pytel, D.; Majsterek, I.; Diehl, J.A. Tumor progression and the different faces of the PERK kinase. Oncogene 2016, 35, 1207–1215. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1α interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef]
- Ali, M.M.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Li, J.; Wang, J.J.; Chen, C.; Tran, J.T.; Saadi, A.; Yu, Q.; Le, Y.Z.; Mandal, M.N.; Anderson, R.E.; et al. X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PLoS ONE 2012, 7, e38616. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Wu, J.; He, G.T.; Zhang, W.J.; Xu, J.; Huang, Q.B. IRE1α Signaling Pathways Involved in Mammalian Cell Fate Determination. Cell. Physiol. Biochem. 2016, 38, 847–858. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef]
- Storniolo, A.; Alfano, V.; Carbotta, S.; Ferretti, E.; Di Renzo, L. IRE1α deficiency promotes tumor cell death and eIF2α degradation through PERK dipendent autophagy. Cell Death Discov. 2018, 4, 3. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Dussmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6α (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jager, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Boil. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Nakano, A. Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via induction of WW domain binding protein 1. J. Biol. Chem. 2011, 286, 35227–35235. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., III; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Pytel, D.; Seyb, K.; Liu, M.; Ray, S.S.; Concannon, J.; Huang, M.; Cuny, G.D.; Diehl, J.A.; Glicksman, M.A. Enzymatic Characterization of ER Stress-Dependent Kinase, PERK, and Development of a High-Throughput Assay for Identification of PERK Inhibitors. J. Biomol. Screen. 2014, 19, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Ji, X.; Liebhaber, S.A.; Diehl, J.A. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2009, 28, 910–920. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [Green Version]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Singleton, D.C.; Harris, A.L. Targeting the ATF4 pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Pike, L.R.; Singleton, D.C.; Buffa, F.; Abramczyk, O.; Phadwal, K.; Li, J.L.; Simon, A.K.; Murray, J.T.; Harris, A.L. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013, 449, 389–400. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, E.; Bal, N.; Kolosov, P. Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity. Int. J. Mol. Sci. 2017, 18, 2213. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Dong, W. Aloe-Emodin Induces Endoplasmic Reticulum Stress-Dependent Apoptosis in Colorectal Cancer Cells. Med. Sci. Monit. 2018, 24, 6331–6339. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef]
- Ibbotson, K.; Yell, J.; Ronaldson, P.T. Nrf2 signaling increases expression of ATP-binding cassette subfamily C mRNA transcripts at the blood-brain barrier following hypoxia-reoxygenation stress. Fluids Barriers CNS 2017, 14, 6. [Google Scholar] [CrossRef]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-B interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Jobbagy, S.; Vitturi, D.A.; Salvatore, S.R.; Turell, L.; Pires, M.F.; Kansanen, E.; Batthyany, C.; Lancaster, J.R., Jr.; Freeman, B.A.; Schopfer, F.J. Electrophiles modulate glutathione reductase activity via alkylation and upregulation of glutathione biosynthesis. Redox Biol. 2019, 21, 101050. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell. Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Mounir, Z.; Krishnamoorthy, J.L.; Wang, S.; Papadopoulou, B.; Campbell, S.; Muller, W.J.; Hatzoglou, M.; Koromilas, A.E. Akt determines cell fate through inhibition of the PERK-eIF2α phosphorylation pathway. Sci. Signal. 2011, 4, ra62. [Google Scholar] [CrossRef]
- Zhang, W.; Hietakangas, V.; Wee, S.; Lim, S.C.; Gunaratne, J.; Cohen, S.M. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013, 27, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. Biomed. Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Corno, C.; Zaffaroni, N.; Perego, P. Role of FoxO Proteins in Cellular Response to Antitumor Agents. Cancers 2019, 11, 90. [Google Scholar] [CrossRef]
- Alasiri, G.; Fan, L.Y.; Zona, S.; Goldsbrough, I.G.; Ke, H.L.; Auner, H.W.; Lam, E.W. ER stress and cancer: The FOXO forkhead transcription factor link. Mol. Cell. Endocrinol. 2018, 462, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [Green Version]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef]
- Jauhiainen, A.; Thomsen, C.; Strombom, L.; Grundevik, P.; Andersson, C.; Danielsson, A.; Andersson, M.K.; Nerman, O.; Rorkvist, L.; Stahlberg, A.; et al. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS ONE 2012, 7, e33208. [Google Scholar] [CrossRef]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fang, H.; Wu, Q.; Wang, X.; Liu, R.; Li, F.; Xiao, J.; Yuan, L.; Zhou, Z.; Ma, J.; et al. Ilamycin E, a natural product of marine actinomycete, inhibits triple-negative breast cancer partially through ER stress-CHOP-Bcl-2. Int. J. Biol. Sci. 2019, 15, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel-Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Investig. 2009, 119, 2925–2941. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tian, B.; Wang, Y.; Ding, H. Kaempferol Sensitizes Human Ovarian Cancer Cells-OVCAR-3 and SKOV-3 to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Apoptosis via JNK/ERK-CHOP Pathway and Up-Regulation of Death Receptors 4 and 5. Med. Sci. Monit. 2017, 23, 5096–5105. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 2009, 284, 1593–1603. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hirata, Y.; Takeda, K.; Hayakawa, Y.; Sato, T.; Kinoshita, H.; Sakamoto, K.; Nakata, W.; Hikiba, Y.; Omata, M.; et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology 2011, 54, 185–195. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Walter, F.; Schmid, J.; Dussmann, H.; Concannon, C.G.; Prehn, J.H. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015, 22, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Lin, Y.; Liu, Y.; You, Y.; Yin, W. IRE1α-TRAF2-ASK1 pathway is involved in CSTMP-induced apoptosis and ER stress in human non-small cell lung cancer A549 cells. Biomed. Pharmacother. 2016, 82, 281–289. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Javelaud, D.; Mauviel, A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment. Cell Melanoma Res. 2008, 21, 429–438. [Google Scholar] [CrossRef]
- Prakasam, A.; Ghose, S.; Oleinik, N.V.; Bethard, J.R.; Peterson, Y.K.; Krupenko, N.I.; Krupenko, S.A. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis. 2014, 5, e1358. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.; Ewings, K.E.; Hadfield, K.; Cook, S.J. Regulatory phosphorylation of Bim: Sorting out the ERK from the JNK. Cell Death Differ. 2005, 12, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schroder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.R.; Phadwal, K.; Simon, A.K.; Harris, A.L. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol. Biol. Rep. 2012, 39, 10811–10822. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Regulated expression of caspase-12 gene in human retinal pigment epithelial cells suggests its immunomodulating role. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5593–5601. [Google Scholar] [CrossRef] [PubMed]
- Labbe, K.; Miu, J.; Yeretssian, G.; Serghides, L.; Tam, M.; Finney, C.A.; Erdman, L.K.; Goulet, M.L.; Kain, K.C.; Stevenson, M.M.; et al. Caspase-12 dampens the immune response to malaria independently of the inflammasome by targeting NF-kappaB signaling. J. Immunol. 2010, 185, 5495–5502. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, M.; Li, J.; Wang, D.; He, Y.; He, J.; Gao, F.; Mai, L.; Li, Y.; Liang, Y.; et al. Activation of UPR Signaling Pathway is Associated with the Malignant Progression and Poor Prognosis in Prostate Cancer. Prostate 2017, 77, 274–281. [Google Scholar] [CrossRef]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef]
- Cuevas, E.P.; Eraso, P.; Mazon, M.J.; Santos, V.; Moreno-Bueno, G.; Cano, A.; Portillo, F. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 2017, 7, 44988. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000, 279, 445–450. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Gazit, G.; Lu, J.; Lee, A.S. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res. Treat. 1999, 54, 135–146. [Google Scholar] [CrossRef]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1263. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Jung, J.H.; Cho, H.M.; Kim, S.H.; Lee, K.M.; Kim, H.J.; Lee, J.H.; Shim, B.Y. GRP78 Protein Expression as Prognostic Values in Neoadjuvant Chemoradiotherapy and Laparoscopic Surgery for Locally Advanced Rectal Cancer. Cancer Res. Treat. 2015, 47, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Banach, A.; Jiang, Y.P.; Roth, E.; Kuscu, C.; Cao, J.; Lin, R.Z. CEMIP upregulates BiP to promote breast cancer cell survival in hypoxia. Oncotarget 2019, 10, 4307–4320. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Alzoubi, K.H.; Khabour, O.F.; Banihani, M.N.; Al-Balas, Q.A.; Swaidan, S. GRP78 regulates sensitivity of human colorectal cancer cells to DNA targeting agents. Cytotechnology 2016, 68, 459–467. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. GRP78 Regulates Apoptosis, Cell Survival and Proliferation in 5-Fluorouracil-resistant SNUC5 Colon Cancer Cells. Anticancer Res. 2017, 37, 4943–4951. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Han, Y.S.; Lee, J.H.; Kim, S.; Lee, S.H. Enhanced Susceptibility to 5-Fluorouracil in Human Colon Cancer Cells by Silencing of GRP78. Anticancer Res. 2017, 37, 2975–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.; Chen, Y.; Huang, S.; Cui, F.; Wang, X. Suppression of GRP78 sensitizes human colorectal cancer cells to oxaliplatin by downregulation of CD24. Oncol. Lett. 2018, 15, 9861–9867. [Google Scholar] [CrossRef] [Green Version]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef]
- Chen, H.Y.; Chang, J.T.; Chien, K.Y.; Lee, Y.S.; You, G.R.; Cheng, A.J. The Endogenous GRP78 Interactome in Human Head and Neck Cancers: A Deterministic Role of Cell Surface GRP78 in Cancer Stemness. Sci. Rep. 2018, 8, 536. [Google Scholar] [CrossRef]
- Wu, M.J.; Jan, C.I.; Tsay, Y.G.; Yu, Y.H.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Chen, Y.S.; Lo, J.F.; Yu, C.C. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol. Cancer 2010, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Benhida, R.; Rocchi, S. Targeting BIP to induce Endoplasmic Reticulum stress and cancer cell death. Oncoscience 2016, 3, 306–307. [Google Scholar] [CrossRef]
- Kosakowska-Cholody, T.; Lin, J.; Srideshikan, S.M.; Scheffer, L.; Tarasova, N.I.; Acharya, J.K. HKH40A downregulates GRP78/BiP expression in cancer cells. Cell Death Dis. 2014, 5, e1240. [Google Scholar] [CrossRef]
- Staquicini, D.I.; D’Angelo, S.; Ferrara, F.; Karjalainen, K.; Sharma, G.; Smith, T.L.; Tarleton, C.A.; Jaalouk, D.E.; Kuniyasu, A.; Baze, W.B.; et al. Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: Preclinical efficacy In Vitro and formal toxicity study of BMTP-78 in rodents and primates. Pharmacogenom. J. 2018, 18, 436–443. [Google Scholar] [CrossRef]
- Zhou, L.; Gao, W.; Wang, K.; Huang, Z.; Zhang, L.; Zhang, Z.; Zhou, J.; Nice, E.C.; Huang, C. Brefeldin A inhibits colorectal cancer growth by triggering Bip/Akt-regulated autophagy. FASEB J. 2019, 33, 5520–5534. [Google Scholar] [CrossRef]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. ER stress and distinct outputs of the IRE1α RNase control proliferation and senescence in response to oncogenic Ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Bujisic, B.; De Gassart, A.; Tallant, R.; Demaria, O.; Zaffalon, L.; Chelbi, S.; Gilliet, M.; Bertoni, F.; Martinon, F. Impairment of both IRE1 expression and XBP1 activation is a hallmark of GCB DLBCL and contributes to tumor growth. Blood 2017, 129, 2420–2428. [Google Scholar] [CrossRef] [Green Version]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Suehara, Y.; Kohsaka, S.; Hayashi, T.; Akaike, K.; Mukaihara, K.; Kurihara, T.; Kim, Y.; Okubo, T.; Ishii, M.; et al. IRE1α-XBP1 inhibitors exerted anti-tumor activities in Ewing’s sarcoma. Oncotarget 2018, 9, 14428–14443. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, F.; Pan, Y.; Chen, X.; Chen, J.; Wang, Y.; Zheng, X.; Zhang, J. XBP1 promotes tumor invasion and is associated with poor prognosis in oral squamous cell carcinoma. Oncol. Rep. 2018, 40, 988–998. [Google Scholar] [CrossRef]
- Xia, T.; Tong, S.; Fan, K.; Zhai, W.; Fang, B.; Wang, S.H.; Wang, J.J. XBP1 induces MMP-9 expression to promote proliferation and invasion in human esophageal squamous cell carcinoma. Am. J. Cancer Res. 2016, 6, 2031–2040. [Google Scholar] [PubMed]
- Pagliarini, V.; Giglio, P.; Bernardoni, P.; De Zio, D.; Fimia, G.M.; Piacentini, M.; Corazzari, M. Downregulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 2015, 128, 1166–1179. [Google Scholar] [CrossRef]
- Yang, J.; Cheng, D.; Zhou, S.; Zhu, B.; Hu, T.; Yang, Q. Overexpression of X-Box Binding Protein 1 (XBP1) Correlates to Poor Prognosis and Up-Regulation of PI3K/mTOR in Human Osteosarcoma. Int. J. Mol. Sci. 2015, 16, 6123. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Jacob, S.; Dunn, B.L.; Georgantopoulos, P.; Zheng, X.L.; Kwaan, H.C.; McKoy, J.M.; Magwood, J.S.; Qureshi, Z.P.; Bandarenko, N.; et al. Ticlopidine-associated ADAMTS13 activity deficient thrombotic thrombocytopenic purpura in 22 persons in Japan: A report from the Southern Network on Adverse Reactions (SONAR). Br. J. Haematol. 2013, 161, 896–898. [Google Scholar] [CrossRef]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.J.; Carlyle, J.R.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, M.; Jin, X.; Ney, G.; Yang, K.B.; Peng, F.; Cao, J.; Iwawaki, T.; Del Valle, J.; Chen, X.; et al. Adaptive endoplasmic reticulum stress signalling via IRE1α-XBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat. Cell Biol. 2019, 21, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Sandoval, T.A.; Chae, C.S.; Chopra, S.; Tan, C.; Rutkowski, M.R.; Raundhal, M.; Chaurio, R.A.; Payne, K.K.; Konrad, C.; et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018, 562, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.A.; Schwarze, S.R. IRE1α controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperon. 2010, 15, 497–508. [Google Scholar] [CrossRef]
- Hu, R.; Warri, A.; Jin, L.; Zwart, A.; Riggins, R.B.; Fang, H.B.; Clarke, R. NF-kappaB signaling is required for XBP1 (unspliced and spliced)-mediated effects on antiestrogen responsiveness and cell fate decisions in breast cancer. Mol. Cell Biol. 2015, 35, 379–390. [Google Scholar] [CrossRef]
- Fang, P.; Xiang, L.; Huang, S.; Jin, L.; Zhou, G.; Zhuge, L.; Li, J.; Fan, H.; Zhou, L.; Pan, C.; et al. IRE1α-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncol. Lett. 2018, 16, 4729–4736. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X. IRE1α-XBP1 pathway promotes melanoma progression by regulating IL-6/STAT3 signaling. J. Transl. Med. 2017, 15, 42. [Google Scholar] [CrossRef]
- Lyu, X.; Zhang, M.; Li, G.; Cai, Y.; Li, G.; Qiao, Q. Interleukin-6 production mediated by the IRE1-XBP1 pathway confers radioresistance in human papillomavirus-negative oropharyngeal carcinoma. Cancer Sci. 2019, 110, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.J.; Yu, H.Q.; Fan, L.L.; Li, X.Q.; Wang, F.; Liu, J.T.; Zhong, F.; Zhang, C.J.; Wei, W.; Wang, H.; et al. Melatonin, a novel selective ATF-6 inhibitor, induces human hepatoma cell apoptosis through COX-2 downregulation. World J. Gastroenterol. 2017, 23, 986–998. [Google Scholar] [CrossRef]
- Ciavattini, A.; Delli Carpini, G.; Serri, M.; Tozzi, A.; Leoni, F.; Di Loreto, E.; Saccucci, F. Unfolded protein response, a link between endometrioid ovarian carcinoma and endometriosis: A pilot study. Oncol. Lett. 2018, 16, 5449–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.C.; Chen, P.C.; Chen, Y.P.; Chang, Y.; Su, I.J. Dominant expression of survival signals of endoplasmic reticulum stress response in Hodgkin lymphoma. Cancer Sci. 2011, 102, 275–281. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells In Vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Spaan, C.N.; Smit, W.L.; van Lidth de Jeude, J.F.; Meijer, B.J.; Muncan, V.; van den Brink, G.R.; Heijmans, J. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis. 2019, 10, 490. [Google Scholar] [CrossRef]
- Hanaoka, M.; Ishikawa, T.; Ishiguro, M.; Tokura, M.; Yamauchi, S.; Kikuchi, A.; Uetake, H.; Yasuno, M.; Kawano, T. Expression of ATF6 as a marker of pre-cancerous atypical change in ulcerative colitis-associated colorectal cancer: A potential role in the management of dysplasia. J. Gastroenterol. 2018, 53, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Hsu, C.C.; Huang, T.T.; Lee, C.H.; Chen, J.L.; Yang, S.H.; Jiang, J.K.; Chen, W.S.; Lee, K.D.; Teng, H.W. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 2018, 12, 1706–1717. [Google Scholar] [CrossRef]
- Coleman, O.I.; Lobner, E.M.; Bierwirth, S.; Sorbie, A.; Waldschmitt, N.; Rath, E.; Berger, E.; Lagkouvardos, I.; Clavel, T.; McCoy, K.D.; et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology 2018, 155, 1539–1552 e1512. [Google Scholar] [CrossRef]
- Wu, X.; Xin, Z.; Zhang, W.; Zheng, S.; Wu, J.; Chen, K.; Wang, H.; Zhu, X.; Li, Z.; Duan, Z.; et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int. J. Cancer 2014, 135, 61–68. [Google Scholar] [CrossRef]
- Papaioannou, A.; Higa, A.; Jegou, G.; Jouan, F.; Pineau, R.; Saas, L.; Avril, T.; Pluquet, O.; Chevet, E. Alterations of EDEM1 functions enhance ATF6 pro-survival signaling. FEBS J. 2018, 285, 4146–4164. [Google Scholar] [CrossRef] [PubMed]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019. [Google Scholar] [CrossRef]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef]
- Yarapureddy, S.; Abril, J.; Foote, J.; Kumar, S.; Asad, O.; Sharath, V.; Faraj, J.; Daniel, D.; Dickman, P.; White-Collins, A.; et al. ATF6α Activation Enhances Survival against Chemotherapy and Serves as a Prognostic Indicator in Osteosarcoma. Neoplasia 2019, 21, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fan, Q.; Quan, H.; Nie, M.; Luo, Y.; Wang, L. The three branches of the unfolded protein response exhibit differential significance in breast cancer growth and stemness. Exp. Cell Res. 2018, 367, 170–185. [Google Scholar] [CrossRef]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Hutzler, M.J.; Wang, S.; Savas, L.; Fraire, A.E. Expression of eukaryotic translation initiation factors 4E and 2α is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer 2001, 92, 2164–2171. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Koifman, L.; Savas, L.; Chen, J.J.; Woda, B.A.; Kadin, M.E. Expression of the translation initiation factors eIF-4E and eIF-2 * is frequently increased in neoplastic cells of Hodgkin lymphoma. Hum. Pathol. 2008, 39, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.V.; Martin, M.E.; Perez, M.I.; Alonso, F.J.; Redondo, C.; Alvarez, M.I.; Salinas, M. Levels, phosphorylation status and cellular localization of translational factor eIF2 in gastrointestinal carcinomas. Histochem. J. 2000, 32, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, I.B.; Wang, S.; Savas, L.; Woda, B.; Pullman, J. Expression of translation initiation factor eIF-2α is increased in benign and malignant melanocytic and colonic epithelial neoplasms. Cancer 2003, 98, 1080–1088. [Google Scholar] [CrossRef]
- Guo, L.; Chi, Y.; Xue, J.; Ma, L.; Shao, Z.; Wu, J. Phosphorylated eIF2α predicts disease-free survival in triple-negative breast cancer patients. Sci. Rep. 2017, 7, 44674. [Google Scholar] [CrossRef]
- Wimbauer, F.; Yang, C.; Shogren, K.L.; Zhang, M.; Goyal, R.; Riester, S.M.; Yaszemski, M.J.; Maran, A. Regulation of interferon pathway in 2-methoxyestradiol-treated osteosarcoma cells. BMC Cancer 2012, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, F.; Xu, J.; Zhang, T.; Wang, G.; Mao, M.; Wang, Z.; Sun, W.; Han, J.; Yang, M.; et al. CYT997 (Lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced ER stress and ROS in osteosarcoma. J. Exp. Clin. Cancer Res. 2019, 38, 44. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Yang, S.F.; Hung, T.W.; Lin, C.L.; Hsieh, Y.H.; Chiou, H.L. Inhibition of eIF2α dephosphorylation accelerates pterostilbene-induced cell death in human hepatocellular carcinoma cells in an ER stress and autophagy-dependent manner. Cell Death Dis. 2019, 10, 418. [Google Scholar] [CrossRef]
- Wang, R.; Sun, D.Z.; Song, C.Q.; Xu, Y.M.; Liu, W.; Liu, Z.; Dong, X.S. Eukaryotic translation initiation factor 2 subunit α (eIF2α) inhibitor salubrinal attenuates paraquat-induced human lung epithelial-like A549 cell apoptosis by regulating the PERK-eIF2α signaling pathway. Toxicol. In Vitro 2018, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK regulates the proliferation and development of insulin-secreting β-cell tumors in the endocrine pancreas of mice. PLoS ONE 2009, 4, e8008. [Google Scholar] [CrossRef]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, S.J.; Ranganathan, A.C.; Adam, A.P.; Iglesias, B.V.; Farias, E.F.; Aguirre-Ghiso, J.A. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS ONE 2007, 2, e615. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2α phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef]
- Pytel, D.; Gao, Y.; Mackiewicz, K.; Katlinskaya, Y.V.; Staschke, K.A.; Paredes, M.C.; Yoshida, A.; Qie, S.; Zhang, G.; Chajewski, O.S.; et al. PERK Is a Haploinsufficient Tumor Suppressor: Gene Dose Determines Tumor-Suppressive Versus Tumor Promoting Properties of PERK in Melanoma. PLoS Genet. 2016, 12, e1006518. [Google Scholar] [CrossRef]
- Aggarwal, P.; Lessie, M.D.; Lin, D.I.; Pontano, L.; Gladden, A.B.; Nuskey, B.; Goradia, A.; Wasik, M.A.; Klein-Szanto, A.J.; Rustgi, A.K.; et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007, 21, 2908–2922. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Ojha, S.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008, 68, 3260–3268. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol 2017, 12, 727–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Tian, B.; Lu, Z.N.; Guo, X.L. Regulation and role of nuclear factor-E2-related factor 2 (Nrf2) in multidrug resistance of hepatocellular carcinoma. Chem. Biol. Interact. 2018, 280, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, W.; Yao, C.; Ren, J.; Zhang, S.; Han, M. Salinomycin overcomes radioresistance in nasopharyngeal carcinoma cells by inhibiting Nrf2 level and promoting ROS generation. Biomed. Pharmacother. 2017, 91, 147–154. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luoni, R.; Ucci, G.; Riccardi, A.; Spriano, P.; Danova, M.; Cassano, E.; Molinari, E.; Ascari, E. Serum thymidine kinase in hematologic malignancies. Haematologica 1988, 73, 31–35. [Google Scholar] [PubMed]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Sci. Rep. 2019, 9, 3210. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Zhang, J.M.; Du, Y.; Wang, J.; Ren, Y.; Li, M.; Li, H.; Cai, Z.; Chu, Q.; Yang, C. Crosstalk between ATF4 and MTA1/HDAC1 promotes osteosarcoma progression. Oncotarget 2016, 7, 7329–7342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallmann, N.; Livgard, M.; Tesikova, M.; Zeynep Nenseth, H.; Akkus, E.; Sikkeland, J.; Jin, Y.; Koc, D.; Kuzu, O.F.; Pradhan, M.; et al. Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Chen, P.; Xi, D.; Zhu, H.; Gao, Y. ATF4 regulates CCL2 expression to promote endometrial cancer growth by controlling macrophage infiltration. Exp. Cell Res. 2017, 360, 105–112. [Google Scholar] [CrossRef]
- Gonzalez-Quiroz, M.; Urra, H.; Limia, C.M.; Hetz, C. Homeostatic interplay between FoxO proteins and ER proteostasis in cancer and other diseases. Semin. Cancer Biol. 2018, 50, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Corno, C.; Stucchi, S.; De Cesare, M.; Carenini, N.; Stamatakos, S.; Ciusani, E.; Minoli, L.; Scanziani, E.; Argueta, C.; Landesman, Y.; et al. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem. Pharmacol. 2018, 147, 93–103. [Google Scholar] [CrossRef]
- Zhao, M.; Luo, R.; Liu, Y.; Gao, L.; Fu, Z.; Fu, Q.; Luo, X.; Chen, Y.; Deng, X.; Liang, Z.; et al. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat. Commun. 2016, 7, 11309. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Mattos, S.; Villalonga, P.; Clardy, J.; Lam, E.W. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol. Cancer Ther. 2008, 7, 3237–3246. [Google Scholar] [CrossRef]
- Ananda Sadagopan, S.K.; Mohebali, N.; Looi, C.Y.; Hasanpourghadi, M.; Pandurangan, A.K.; Arya, A.; Karimian, H.; Mustafa, M.R. Forkhead Box Transcription Factor (FOXO3a) mediates the cytotoxic effect of vernodalin In Vitro and inhibits the breast tumor growth In Vivo. J. Exp. Clin. Cancer Res. 2015, 34, 147. [Google Scholar] [CrossRef]
- Liu, H.; Yin, J.; Wang, C.; Gu, Y.; Deng, M.; He, Z. FOXO3a mediates the cytotoxic effects of cisplatin in lung cancer cells. Anticancer Drugs 2014, 25, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Zhang, L.; Gong, Z.; Hou, X.; Xu, Y.; Feng, X.; Wang, H.; You, H. FoxO3 inactivation promotes human cholangiocarcinoma tumorigenesis and chemoresistance through Keap1-Nrf2 signaling. Hepatology 2016, 63, 1914–1927. [Google Scholar] [CrossRef] [Green Version]
- Alasiri, G.; Jiramongkol, Y.; Zona, S.; Fan, L.Y.; Mahmud, Z.; Gong, G.; Lee, H.J.; Lam, E.W. Regulation of PERK expression by FOXO3: A vulnerability of drug-resistant cancer cells. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, P.; Klotz, L.O. Posttranscriptional regulation of FOXO expression: microRNAs and beyond. Br. J. Pharmacol. 2017, 174, 1514–1532. [Google Scholar] [CrossRef]
- Niyonizigiye, I.; Ngabire, D.; Patil, M.P.; Singh, A.A.; Kim, G.D. In Vitro induction of endoplasmic reticulum stress in human cervical adenocarcinoma HeLa cells by fucoidan. Int. J. Biol. Macromol. 2019, 137, 844–852. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef]
- DeZwaan-McCabe, D.; Riordan, J.D.; Arensdorf, A.M.; Icardi, M.S.; Dupuy, A.J.; Rutkowski, D.T. The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 2013, 9, e1003937. [Google Scholar] [CrossRef]
- Cao, Y.; Trillo-Tinoco, J.; Sierra, R.A.; Anadon, C.; Dai, W.; Mohamed, E.; Cen, L.; Costich, T.L.; Magliocco, A.; Marchion, D.; et al. ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat. Commun. 2019, 10, 1280. [Google Scholar] [CrossRef] [PubMed]
- Girnius, N.; Edwards, Y.J.; Garlick, D.S.; Davis, R.J. The cJUN NH2-terminal kinase (JNK) signaling pathway promotes genome stability and prevents tumor initiation. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Insua-Rodriguez, J.; Pein, M.; Hongu, T.; Meier, J.; Descot, A.; Lowy, C.M.; De Braekeleer, E.; Sinn, H.P.; Spaich, S.; Sutterlin, M.; et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Xie, X.; Kaoud, T.S.; Edupuganti, R.; Zhang, T.; Kogawa, T.; Zhao, Y.; Chauhan, G.B.; Giannoukos, D.N.; Qi, Y.; Tripathy, D.; et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene 2017, 36, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; An, G.; Kuo, M.T. C-Jun N-terminal kinase signalling pathway in response to cisplatin. J. Cell. Mol. Med. 2016, 20, 2013–2019. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Pilling, A.B.; Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar] [CrossRef]
- Wolf, P. BH3 Mimetics for the Treatment of Prostate Cancer. Front. Pharmacol. 2017, 8, 557. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef]
- Liu, Q.; Osterlund, E.J.; Chi, X.; Pogmore, J.; Leber, B.; Andrews, D.W. Bim escapes displacement by BH3-mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. Elife 2019, 8. [Google Scholar] [CrossRef]
- Chu, W.K.; Hsu, C.C.; Huang, S.F.; Hsu, C.C.; Chow, S.E. Caspase 12 degrades IkBα protein and enhances MMP-9 expression in human nasopharyngeal carcinoma cell invasion. Oncotarget 2017, 8, 33515–33526. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Dourdin, N.; Wu, C.; De Veyra, T.; Elce, J.S.; Greer, P.A. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2006, 281, 16016–16024. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, Z.; Wang, C.; Zhou, H.; Liu, W.; Kanchana, K.; Dai, X.; Zou, P.; Gu, J.; Cai, L.; et al. Curcumin derivative WZ35 efficiently suppresses colon cancer progression through inducing ROS production and ER stress-dependent apoptosis. Am. J. Cancer Res. 2017, 7, 275–288. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Denning, T.L.; Takaishi, H.; Crowe, S.E.; Boldogh, I.; Jevnikar, A.; Ernst, P.B. Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells. Free Radic. Biol. Med. 2002, 33, 1641–1650. [Google Scholar] [CrossRef]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993, 12, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Regulated necrotic cell death: The passive aggressive side of Bax and Bak. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, M.; Yang, Z.; Wang, W.; Lin, H.; Xu, S. Selenoprotein S silencing triggers mouse hepatoma cells apoptosis and necrosis involving in intracellular calcium imbalance and ROS-mPTP-ATP. Biochim Biophys. Acta Gen. Subj. 2018, 1862, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.W.; Cho, H.S.; Kim, S.H.; Kim, W.J.; Jung, J.Y. Ascorbic Acid Induces Necrosis in Human Laryngeal Squamous Cell Carcinoma via ROS, PKC, and Calcium Signaling. J. Cell. Physiol. 2017, 232, 417–425. [Google Scholar] [CrossRef]
- Levine, A.S.; Sun, L.; Tan, R.; Gao, Y.; Yang, L.; Chen, H.; Teng, Y.; Lan, L. The oxidative DNA damage response: A review of research undertaken with Tsinghua and Xiangya students at the University of Pittsburgh. Sci. China Life Sci. 2017, 60, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Brewer, T.F.; Garcia, F.J.; Onak, C.S.; Carroll, K.S.; Chang, C.J. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 2015, 84, 765–790. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Jiang, X.; Zhao, C.; Lee, C.; Ferriero, D.M. Exogenous low dose hydrogen peroxide increases hypoxia-inducible factor-1α protein expression and induces preconditioning protection against ischemia in primary cortical neurons. Neurosci. Lett. 2008, 441, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Smith, A.L.; Andrews, K.L.; Beckmann, H.; Bellon, S.F.; Beltran, P.J.; Booker, S.; Chen, H.; Chung, Y.A.; D’Angelo, N.D.; Dao, J.; et al. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2015, 58, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Chien, C.C.; Chen, Y.C.; Chiu, W.T. Protein Kinase RNA-Like Endoplasmic Reticulum Kinase-Mediated Bcl-2 Protein Phosphorylation Contributes to Evodiamine-Induced Apoptosis of Human Renal Cell Carcinoma Cells. PLoS ONE 2016, 11, e0160484. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Dziki, L.; Nowak, A.; Dziki, A.; Diehl, J.A.; Majsterek, I. Inhibition of PERK-dependent pro-adaptive signaling pathway as a promising approach for cancer treatment. Pol. Prz. Chir. 2017, 89, 7–10. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Soto, P.; Lopez, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Chien, C.C.; Wu, M.S.; Chen, Y.C. Evodiamine from Evodia rutaecarpa induces apoptosis via activation of JNK and PERK in human ovarian cancer cells. Phytomedicine 2016, 23, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, M.; Zou, P.; Kanchana, K.; Weng, Q.; Chen, W.; Zhong, P.; Ji, J.; Zhou, H.; He, L.; et al. Curcumin analog WZ35 induced cell death via ROS-dependent ER stress and G2/M cell cycle arrest in human prostate cancer cells. BMC Cancer 2015, 15, 866. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Al-Alem, U.; Ali, M.M.; Bosland, M.C. Genistein increases estrogen receptor β expression in prostate cancer via reducing its promoter methylation. J. Steroid Biochem. Mol. Biol. 2015, 152, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fan, J.; Cheng, L.; Hu, P.; Liu, R. The anticancer activity of genistein is increased in estrogen receptor β 1-positive breast cancer cells. Onco Targets Ther. 2018, 11, 8153–8163. [Google Scholar] [CrossRef]
- Wei, H.; Wei, L.; Frenkel, K.; Bowen, R.; Barnes, S. Inhibition of tumor promoter-induced hydrogen peroxide formation In Vitro and In Vivo by genistein. Nutr. Cancer 1993, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bowen, R.; Cai, Q.; Barnes, S.; Wang, Y. Antioxidant and antipromotional effects of the soybean isoflavone genistein. Proc. Soc. Exp. Biol. Med. 1995, 208, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.L.; Min, M.; Shen, W.; Liu, Y. Genistein induced anticancer effects on pancreatic cancer cell lines involves mitochondrial apoptosis, G0/G1cell cycle arrest and regulation of STAT3 signalling pathway. Phytomedicine 2018, 39, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bao, J.; Yang, J. Genistein-triggered anticancer activity against liver cancer cell line HepG2 involves ROS generation, mitochondrial apoptosis, G2/M cell cycle arrest and inhibition of cell migration. Arch. Med. Sci. 2019, 15, 1001–1009. [Google Scholar] [CrossRef]
- Susanikova, I.; Puchl’ova, M.; Lachova, V.; Svajdlenka, E.; Mucaji, P.; Smetana, K., Jr.; Gal, P. Genistein and Selected Phytoestrogen-Containing Extracts Differently Modulate Antioxidant Properties and Cell Differentiation: An In Vitro Study in NIH-3T3, HaCaT and MCF-7 Cells. Folia Biol. 2019, 65, 24–35. [Google Scholar]
- Kim, I.G.; Kim, J.S.; Lee, J.H.; Cho, E.W. Genistein decreases cellular redox potential, partially suppresses cell growth in HL60 leukemia cells and sensitizes cells to γ-radiation-induced cell death. Mol. Med. Rep. 2014, 10, 2786–2792. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; et al. Genistein and cancer: Current status, challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef]
- Hsiao, Y.C.; Peng, S.F.; Lai, K.C.; Liao, C.L.; Huang, Y.P.; Lin, C.C.; Lin, M.L.; Liu, K.C.; Tsai, C.C.; Ma, Y.S.; et al. Genistein induces apoptosis In Vitro and has antitumor activity against human leukemia HL-60 cancer cell xenograft growth In Vivo. Environ. Toxicol. 2019, 34, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jager, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.C.; Adam, A.P.; Zhang, L.; Aguirre-Ghiso, J.A. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biol. Ther. 2006, 5, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, T.; Odisho, T.; Sidorova, E.; Volchuk, A. Pancreatic β-cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell Physiol. 2012, 302, C992–C1003. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. https://doi.org/10.3390/ijms20184354
Siwecka N, Rozpędek W, Pytel D, Wawrzynkiewicz A, Dziki A, Dziki Ł, Diehl JA, Majsterek I. Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. International Journal of Molecular Sciences. 2019; 20(18):4354. https://doi.org/10.3390/ijms20184354
Chicago/Turabian StyleSiwecka, Natalia, Wioletta Rozpędek, Dariusz Pytel, Adam Wawrzynkiewicz, Adam Dziki, Łukasz Dziki, J. Alan Diehl, and Ireneusz Majsterek. 2019. "Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis" International Journal of Molecular Sciences 20, no. 18: 4354. https://doi.org/10.3390/ijms20184354