Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
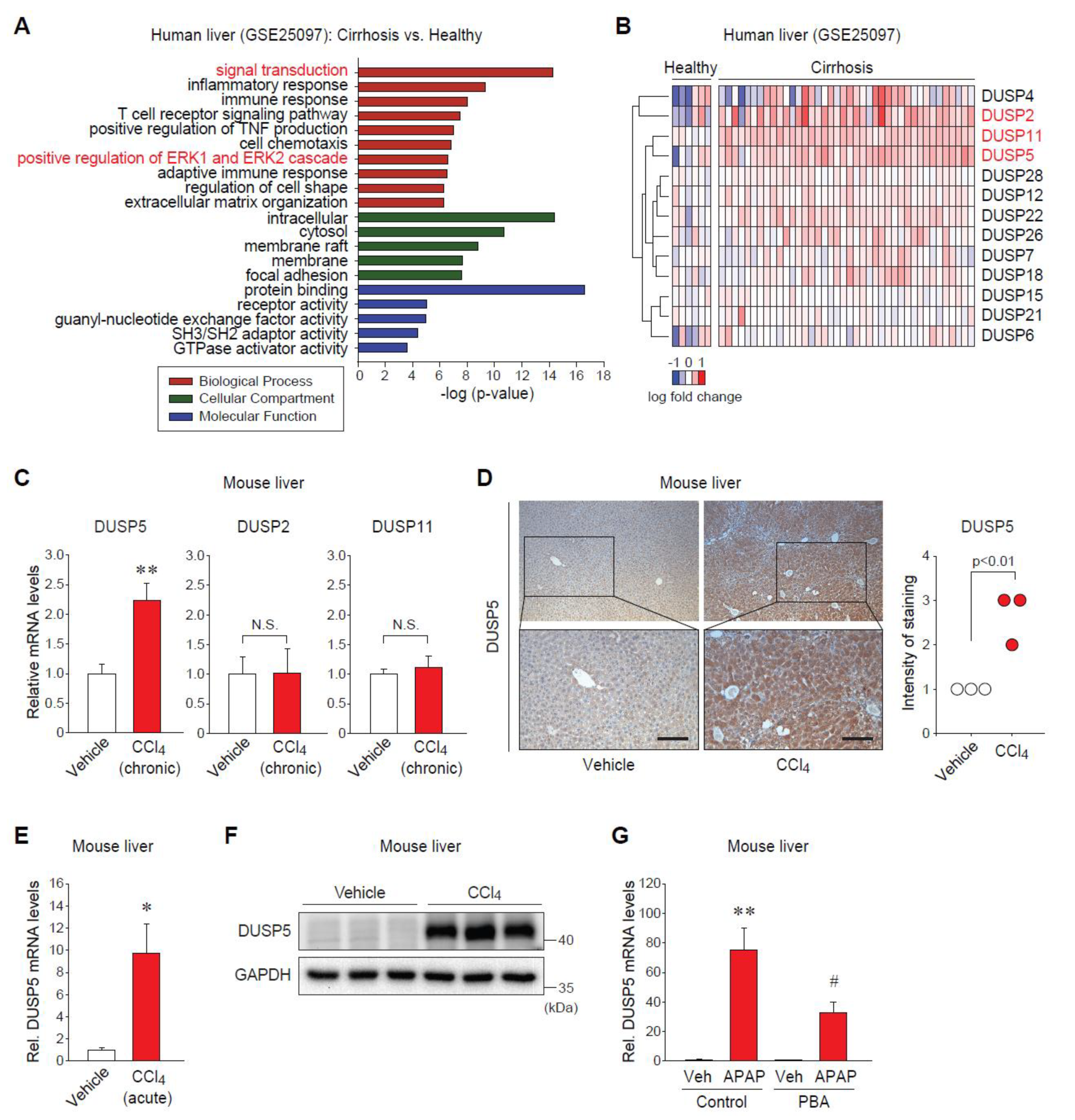
2.1. Dual-Specificity Phosphatase 5 (DUSP5) Expression Is Elevated in Patients and Mice with Liver Diseases
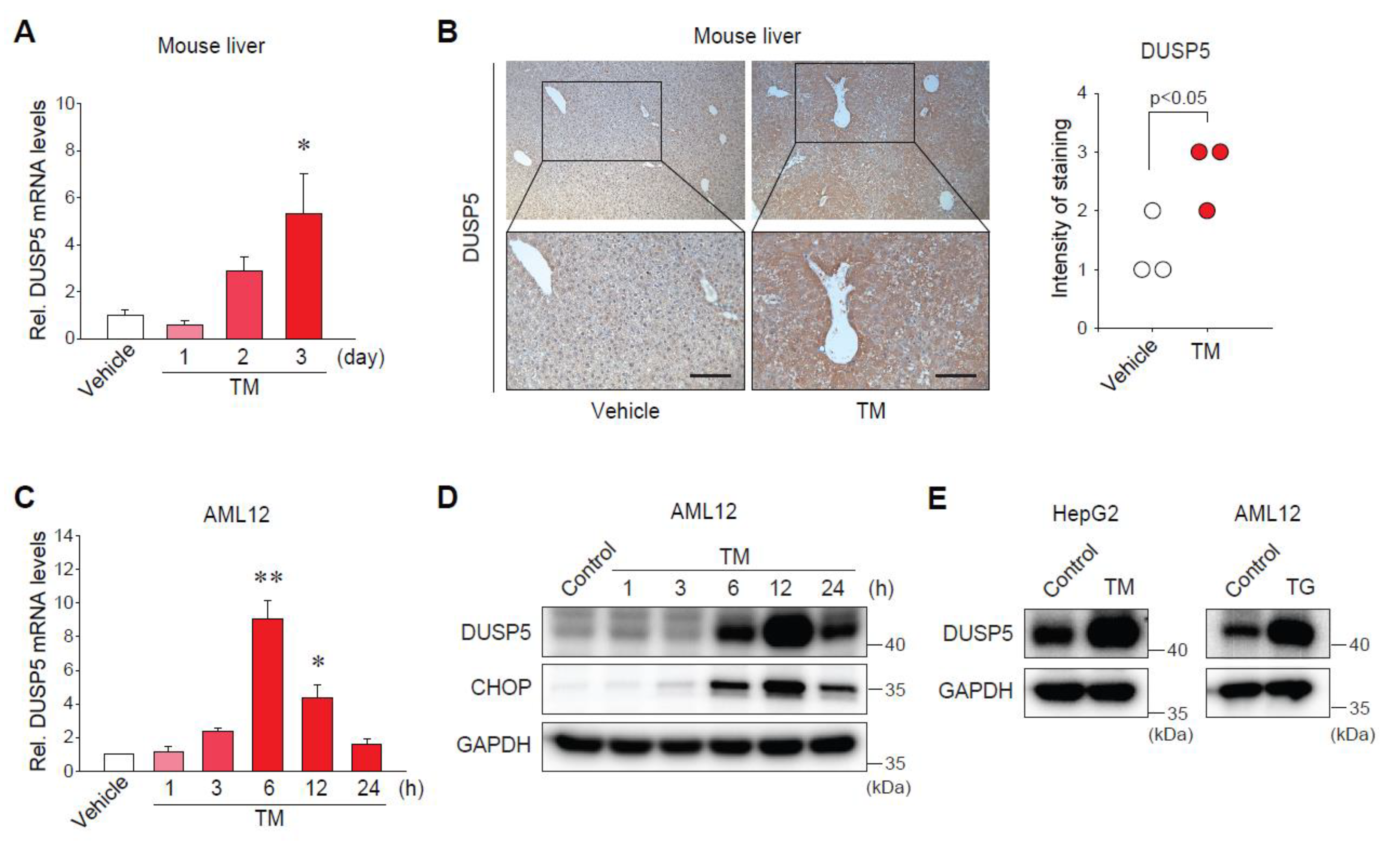
2.2. Endoplasmic Reticulum (ER) Stress Increases DUSP5 Expression in Hepatocytes
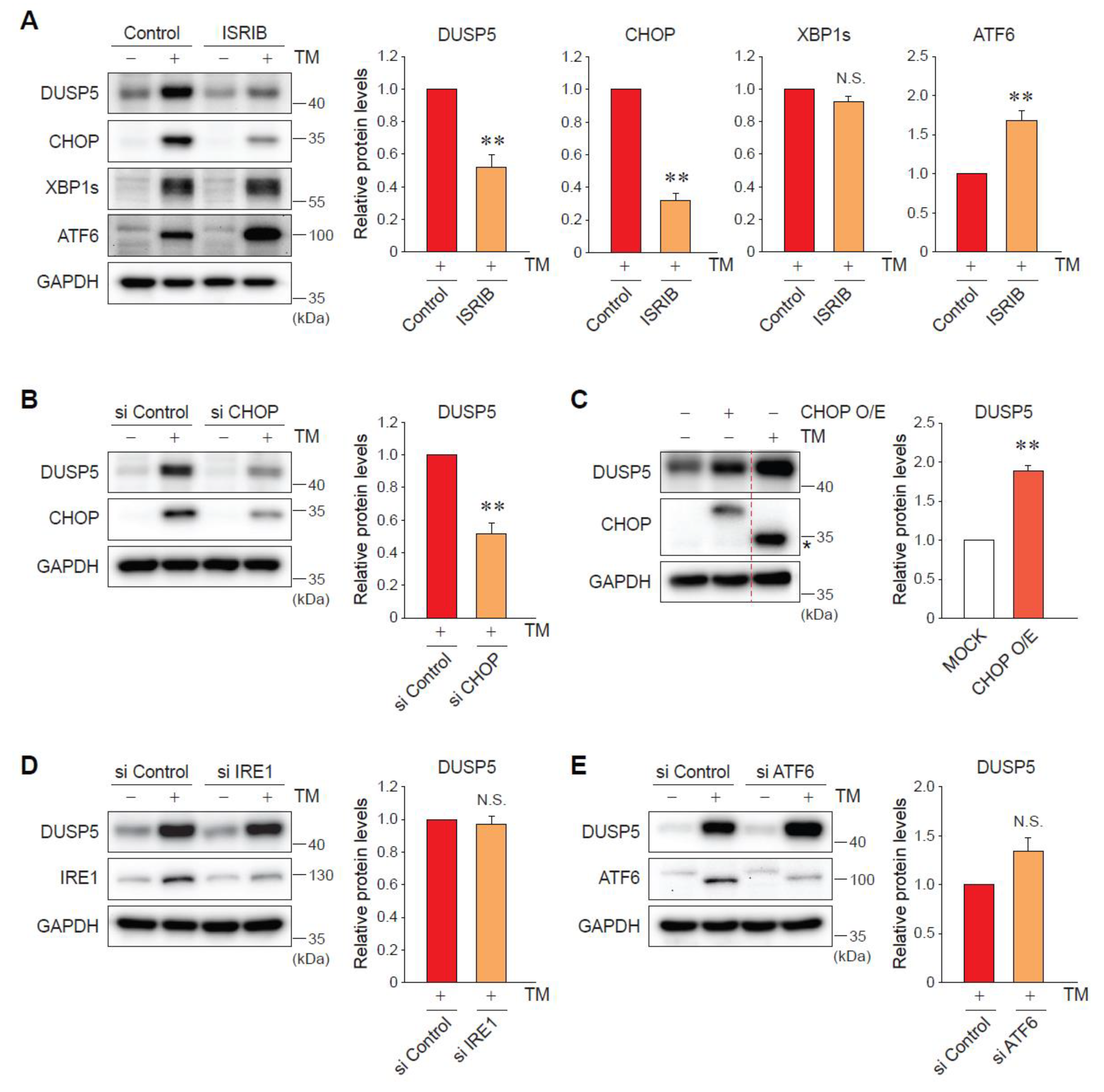
2.3. ER Stress-Induced DUSP5 Expression Is Mediated by the Protein Kinase RNA-Like Endoplasmic Reticulum Kinase (PERK)-C/EBP Homologous Protein (CHOP) Pathway
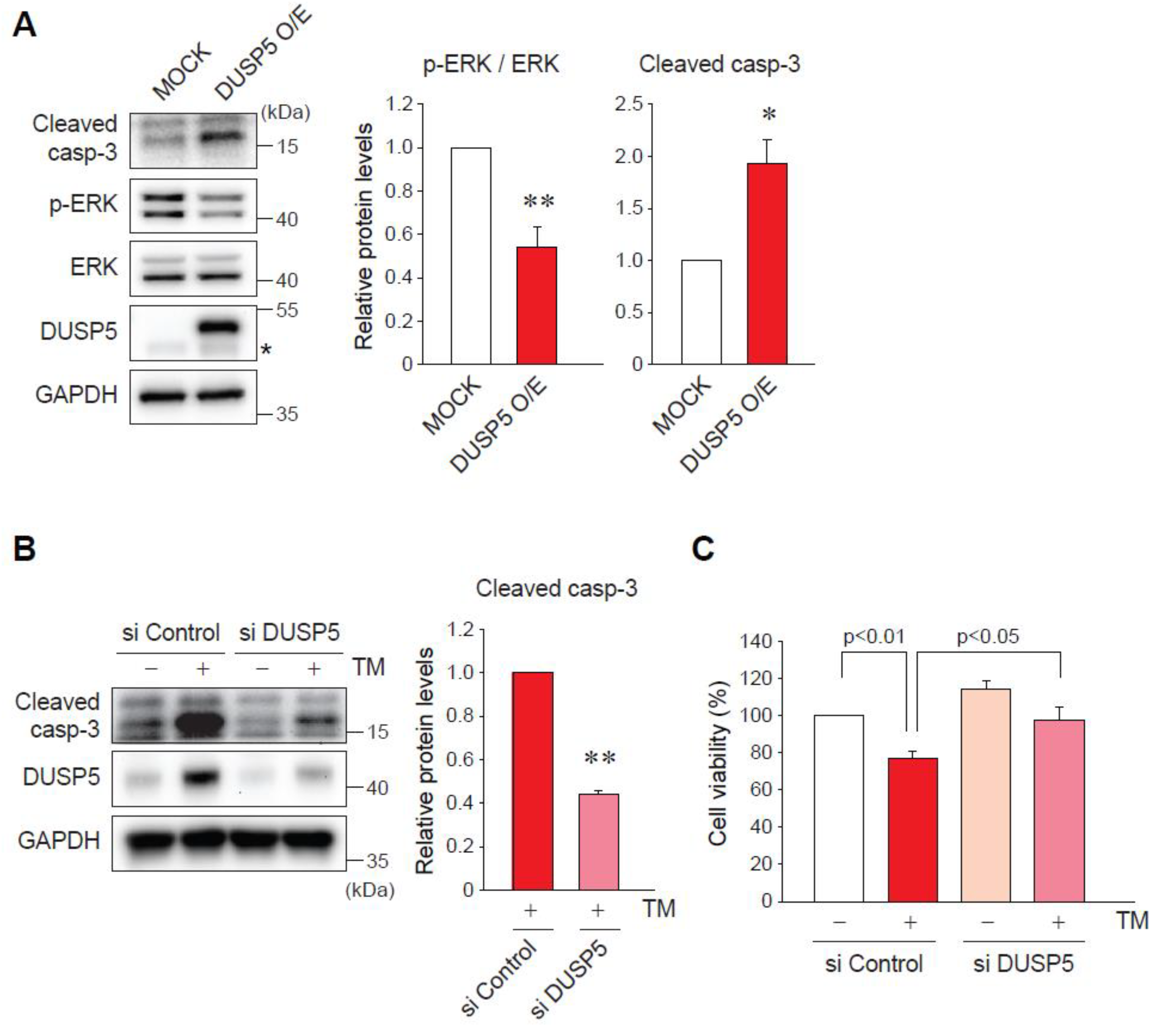
2.4. DUSP5 Overexpression by ER Stress Induces Hepatocyte Death
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bioinformatic Analysis
4.3. Animal Treatments
4.4. RNA Isolation and Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Assays
4.5. Immunohistochemistry
4.6. Cell Culture
4.7. Immunoblottings
4.8. Transient Transfection and Small Interfering RNA (siRNA) Knockdown
4.9. Methylthiazolyldiphenyl-tetrazolium bromide (MTT) Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APAP | Acetaminophen |
| ATF6 | Activating transcription factor 6 |
| CCl4 | Carbon tetrachloride |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| DUSP | Dual-specificity phosphatase |
| ERK | Extracellular-signal-regulated kinase |
| ER stress | Endoplasmic reticulum stress |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GEO | Gene Expression Omnibus |
| GO | Gene ontology |
| IRE1 | Inositol-requiring enzyme 1 |
| MAPK | Mitogen-activated protein kinase |
| PBA | 4-phenylbutyrate |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| TG | Thapsigargin |
| TM | Tunicamycin |
| UPR | Unfolded protein response |
| XBP1 | X-box-binding protein-1 |
References
- Malhi, H.; Gores, G.J. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef]
- Wattacheril, J.; Issa, D.; Sanyal, A. Nonalcoholic Steatohepatitis (NASH) and hepatic fibrosis: Emerging therapies. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 649–662. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of dual-specificity phosphatase (DUSP) ubiquitination and protein stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Lang, R.; Raffi, F.A.M. Dual-specificity phosphatases in immunity and infection: An update. Int. J. Mol. Sci. 2019, 20, 2710. [Google Scholar] [CrossRef]
- Han, C.Y.; Lim, S.W.; Koo, J.H.; Kim, W.; Kim, S.G. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut 2016, 65, 1377–1388. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Mandl, M.; Slack, D.N.; Keyse, S.M. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol. Cell Biol. 2005, 25, 1830–1845. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Ye, P.; Xiang, M.; Liao, H.; Liu, J.; Luo, H.; Wang, Y.; Huang, L.; Chen, M.; Xia, J. Dual-specificity phosphatase 9 protects against nonalcoholic fatty liver disease in mice through ASK1 suppression. Hepatology 2019, 69, 76–93. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, L.M.; Zhang, J.L.; Sabri, A.; Wang, S.J.; Qin, G.J.; Guo, C.Q.; Wen, H.T.; Du, B.B.; Zhang, D.H.; et al. Dual specificity phosphatase 12 regulates hepatic lipid metabolism through inhibition of the lipogenesis and apoptosis signal-regulating kinase 1 pathways. Hepatology 2019. [Google Scholar] [CrossRef]
- Ye, P.; Liu, J.; Xu, W.; Liu, D.; Ding, X.; Le, S.; Zhang, H.; Chen, S.; Chen, M.; Xia, J. Dual-specificity phosphatase 26 protects against nonalcoholic fatty liver disease in mice through transforming growth factor beta-activated kinase 1 suppression. Hepatology 2019, 69, 1946–1964. [Google Scholar] [CrossRef]
- Buffet, C.; Catelli, M.G.; Hecale-Perlemoine, K.; Bricaire, L.; Garcia, C.; Gallet-Dierick, A.; Rodriguez, S.; Cormier, F.; Groussin, L. Dual specificity phosphatase 5, a specific negative regulator of ERK signaling, is Induced by serum response factor and Elk-1 transcription factor. PLoS ONE 2015, 10, e0145484. [Google Scholar] [CrossRef]
- Du, M.; Zhuang, Y.; Tan, P.; Yu, Z.; Zhang, X.; Wang, A. microRNA-95 knockdown inhibits epithelial-mesenchymal transition and cancer stem cell phenotype in gastric cancer cells through MAPK pathway by upregulating DUSP5. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Yan, T.; Zhang, F.; Sun, C.; Sun, J.; Wang, Y.; Xu, X.; Shi, J.; Shi, G. miR-32-5p-mediated Dusp5 downregulation contributes to neuropathic pain. Biochem. Biophys. Res. Commun. 2018, 495, 506–511. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef]
- Kucharska, A.; Rushworth, L.K.; Staples, C.; Morrice, N.A.; Keyse, S.M. Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell. Signal. 2009, 21, 1794–1805. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Zhang, C.; He, X.; Murphy, S.R.; Zhang, H.; Wang, S.; Ge, Y.; Gao, W.; Williams, J.M.; Geurts, A.M.; Roman, R.J.; et al. Knockout of dual-specificity protein phosphatase 5 protects against hypertension-induced renal injury. J. Pharmacol. Exp. Ther. 2019, 370, 206–217. [Google Scholar] [CrossRef]
- Seo, H.; Cho, Y.C.; Ju, A.; Lee, S.; Park, B.C.; Park, S.G.; Kim, J.H.; Kim, K.; Cho, S. Dual-specificity phosphatase 5 acts as an anti-inflammatory regulator by inhibiting the ERK and NF-kappaB signaling pathways. Sci. Rep. 2017, 7, 17348. [Google Scholar] [CrossRef]
- Han, C.Y.; Koo, J.H.; Kim, S.H.; Gardenghi, S.; Rivella, S.; Strnad, P.; Hwang, S.J.; Kim, S.G. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat. Commun. 2016, 7, 13817. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.-S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. https://doi.org/10.3390/ijms20184369
Jo HJ, Yang JW, Park JH, Choi ES, Lim C-S, Lee S, Han CY. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. International Journal of Molecular Sciences. 2019; 20(18):4369. https://doi.org/10.3390/ijms20184369
Chicago/Turabian StyleJo, Hye Jin, Jin Won Yang, Ji Hye Park, Eul Sig Choi, Chae-Seok Lim, Seoul Lee, and Chang Yeob Han. 2019. "Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death" International Journal of Molecular Sciences 20, no. 18: 4369. https://doi.org/10.3390/ijms20184369
APA StyleJo, H. J., Yang, J. W., Park, J. H., Choi, E. S., Lim, C.-S., Lee, S., & Han, C. Y. (2019). Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. International Journal of Molecular Sciences, 20(18), 4369. https://doi.org/10.3390/ijms20184369