FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic ? Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
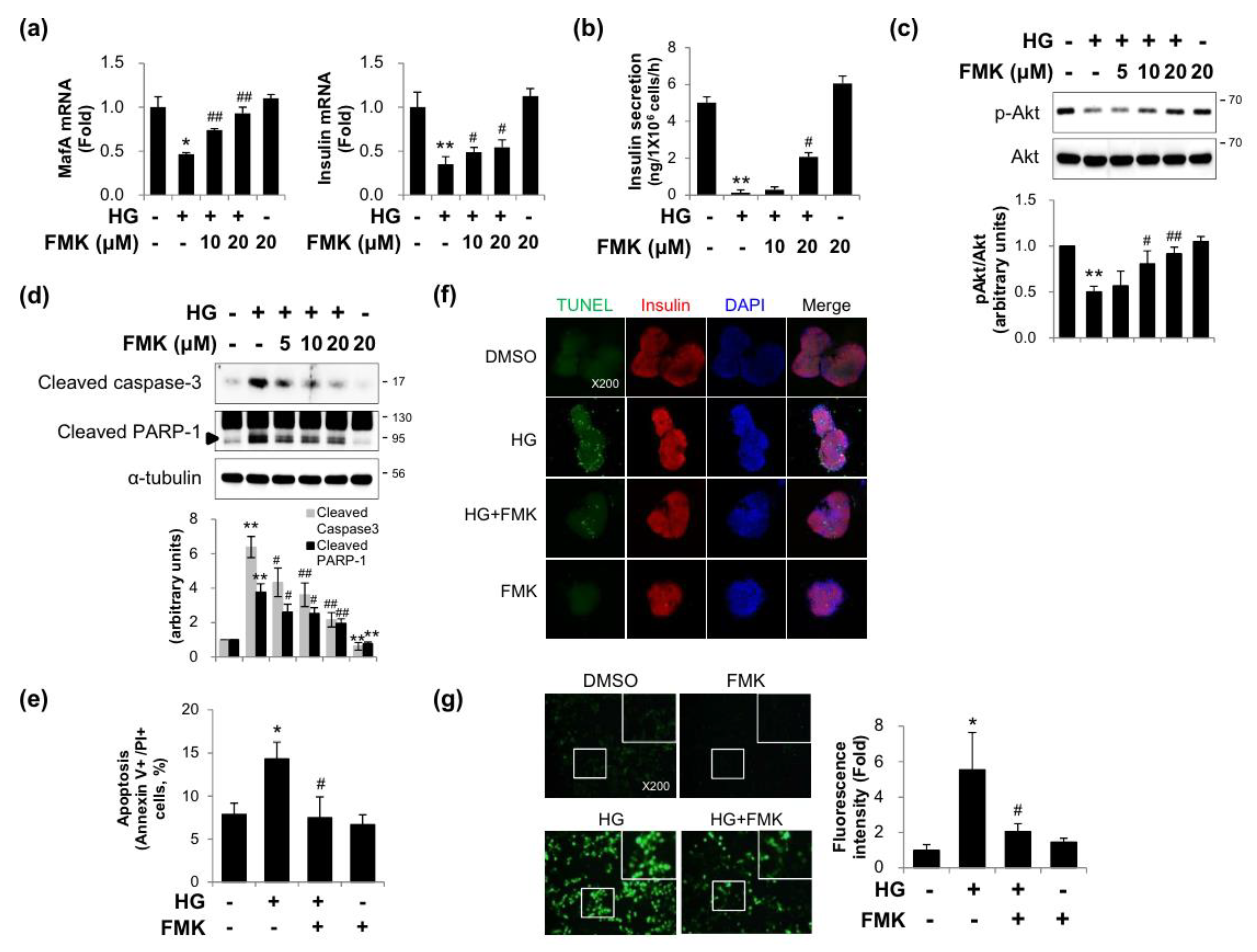
2.1. FMK Protected INS-1 Cells from High Glucose-Induced β-cell Dysfunction, Apoptosis, and Oxidative Stress
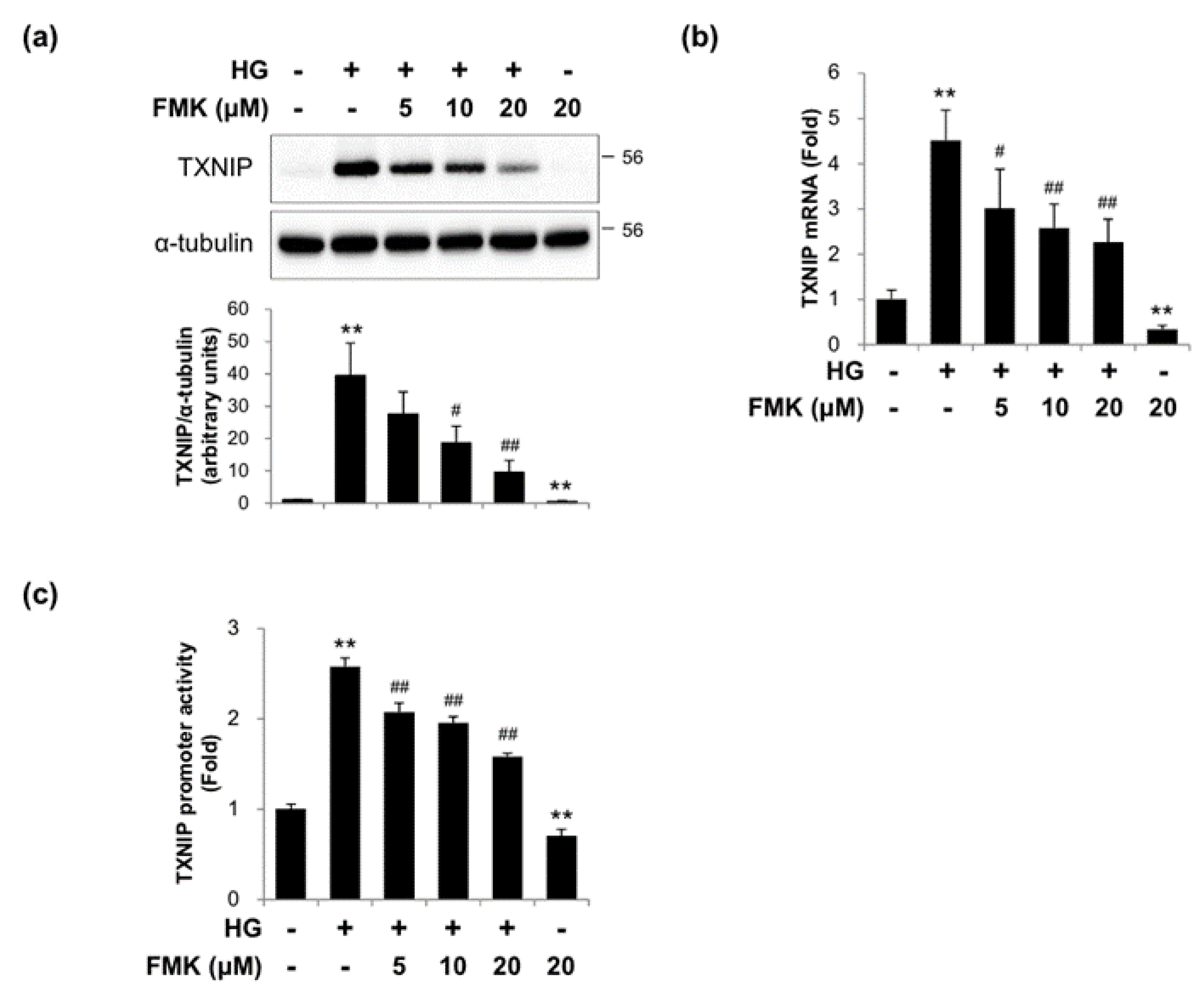
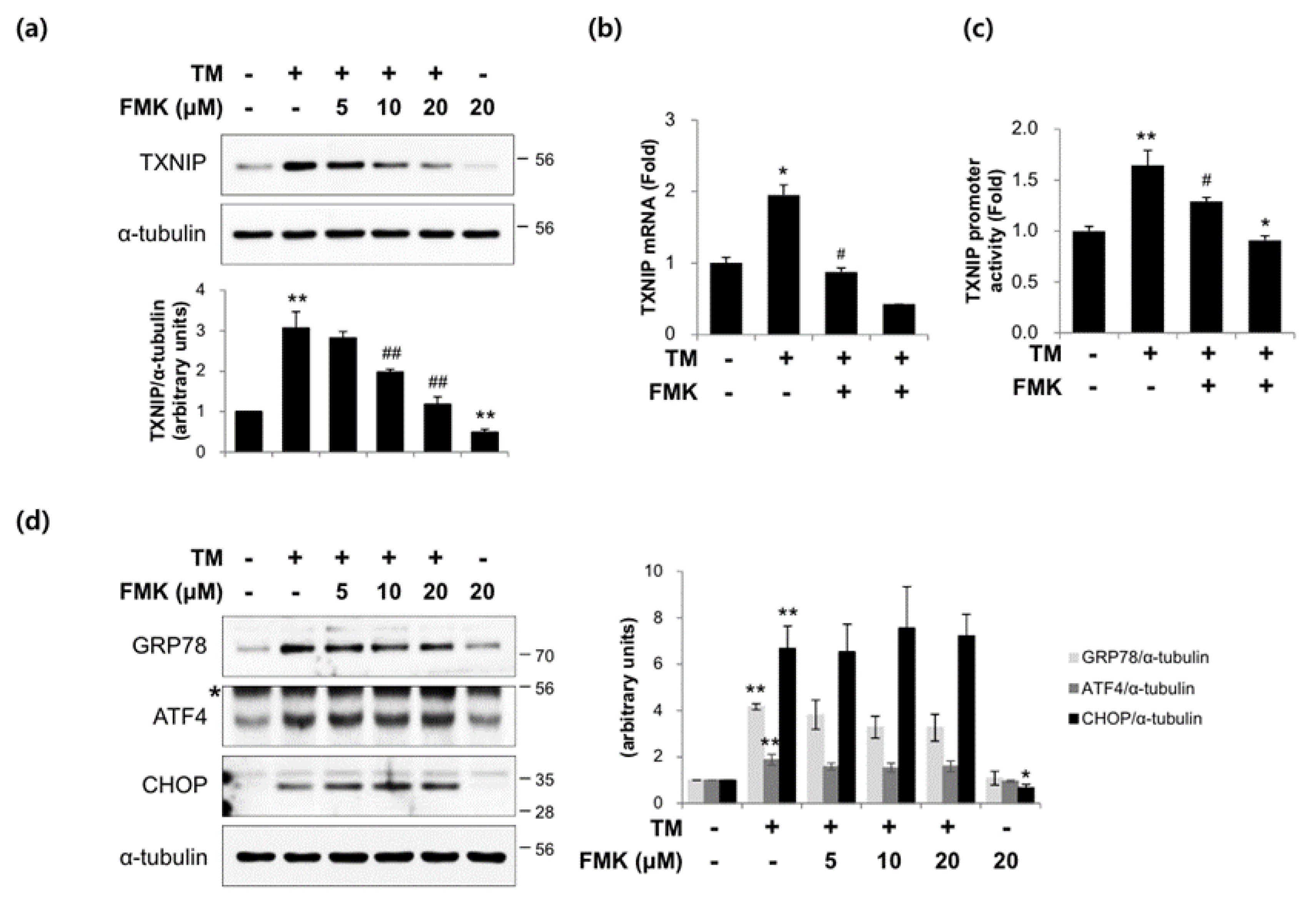
2.2. FMK Inhibited High Glucose-Induced TXNIP Expression in INS-1 Cells
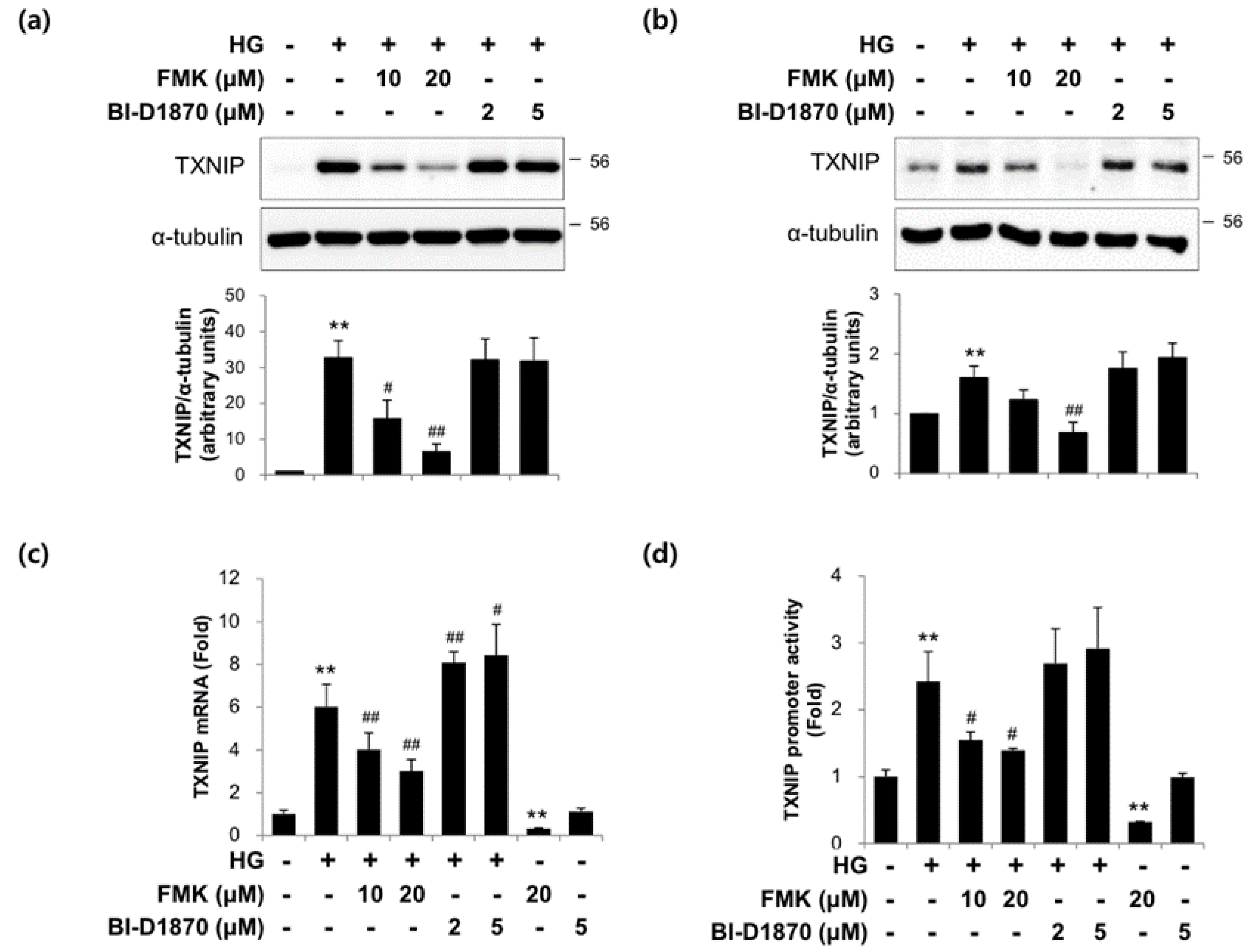
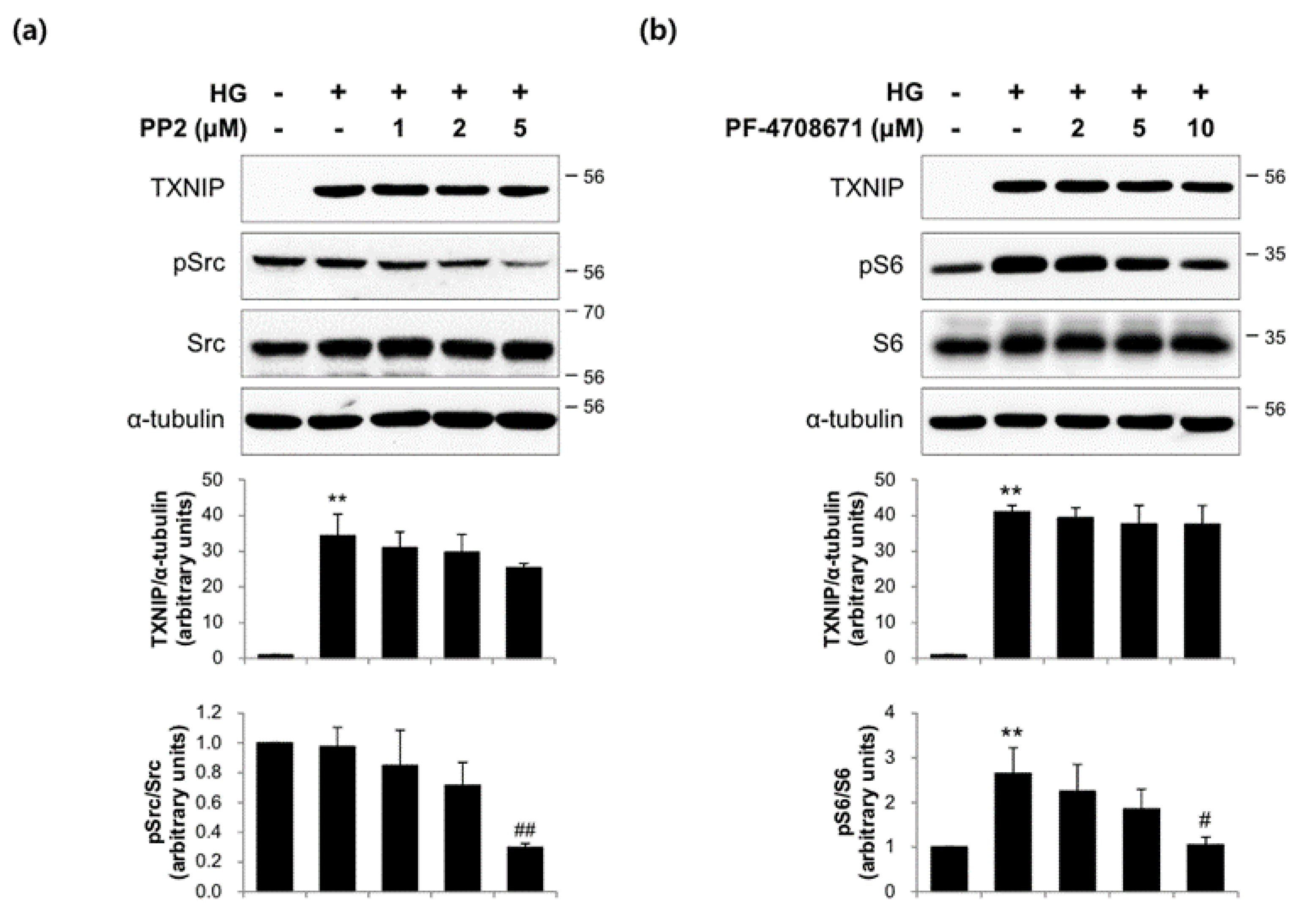
2.3. The Actions of FMK Are Not Mediated by p90RSK, Src, or S6K1 Kinases in INS-1 Cells
2.4. FMK Inhibited the HG-Induced Nuclear Translocation and Activity of ChREBP in INS-1 Cells
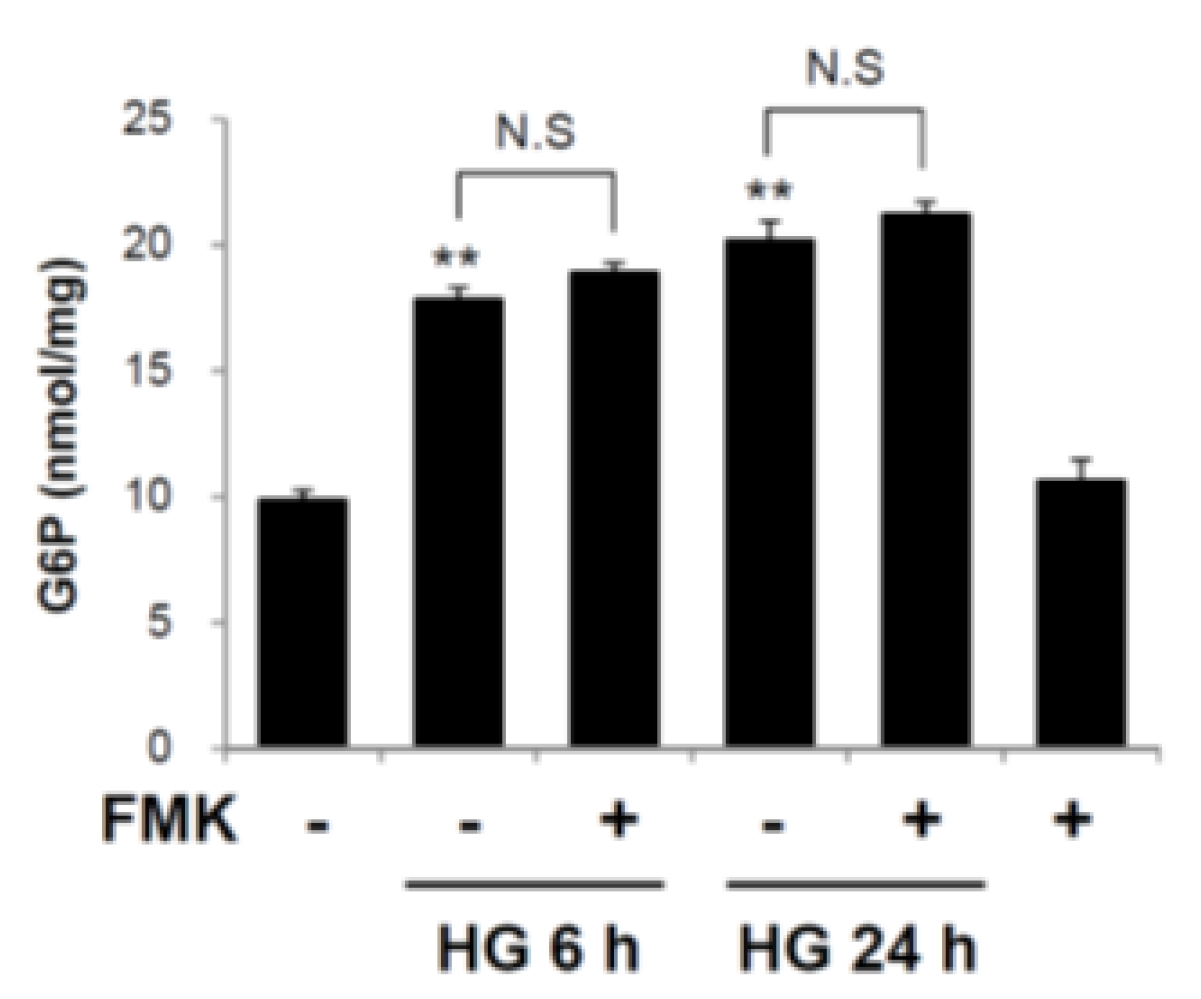
2.5. Glucose 6-Phosphate is not a Target of FMK-Mediated ChREBP Regulation
2.6. FMK Inhibited Tunicamycin-Induced TXNIP Expression in INS-1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Islet Isolation
4.3. Plasmid DNA Transfection and Reporter Gene Assay
4.4. Small Interfering RNA (siRNA)
4.5. Quantitative Real Time RT-PCR (qRT-PCR)
4.6. Western Blotting
4.7. Subcellular Fractionation
4.8. Measurement of Intracellular Reactive Oxygen Species (ROS)
4.9. Immunofluorescence Imaging
4.10. TUNEL Assay
4.11. Glucose Sitmulated Insulin Secretion (GSIS)
4.12. Flow Cytometry
4.13. Measurement of Glucose-6-Phosphate (G6P)
4.14. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TXNIP | Thioredoxin-interacting protein |
| ChREBP | Carbohydrate response element-binding protein |
| HG | High glucose |
| TM | Tunicamycin |
References
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: From mechanisms to evaluation and clinical implications. Biochem. Med. (Zagreb) 2013, 23, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Gu, J.; Meng, X.; Zheng, H.; Wang, D. Systematic investigation into the role of intermittent high glucose in pancreatic beta-cells. Int. J. Clin. Exp. Med. 2015, 8, 5462–5469. [Google Scholar] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54 (Suppl. 2), S108–S113. [Google Scholar] [CrossRef]
- Wang, H.; Kouri, G.; Wollheim, C.B. Er stress and srebp-1 activation are implicated in beta-cell glucolipotoxicity. J. Cell Sci. 2005, 118, 3905–3915. [Google Scholar] [CrossRef]
- Oh, Y.S.; Lee, Y.J.; Kang, Y.; Han, J.; Lim, O.K.; Jun, H.S. Exendin-4 inhibits glucolipotoxic er stress in pancreatic beta cells via regulation of srebp1c and c/ebpbeta transcription factors. J. Endocrinol. 2013, 216, 343–352. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin d(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [PubMed]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid Redox. Signal 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Kim, S.Y.; Suh, H.W.; Chung, J.W.; Yoon, S.R.; Choi, I. Diverse functions of vdup1 in cell proliferation, differentiation, and diseases. Cell Mol. Immunol. 2007, 4, 345–351. [Google Scholar]
- Wang, W.; Wang, C.; Ding, X.Q.; Pan, Y.; Gu, T.T.; Wang, M.X.; Liu, Y.L.; Wang, F.M.; Wang, S.J.; Kong, L.D. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. Br. J. Pharmacol. 2013, 169, 1352–1371. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.J. Roles of thioredoxin binding protein (txnip) in oxidative stress, apoptosis and cancer. Mitochondrion 2013, 13, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A.; Pise-Masison, C.A.; Radonovich, M.; Hoffmann, S.C.; Hirshberg, B.; Brady, J.N.; Harlan, D.M. Oligonucleotide microarray analysis of intact human pancreatic islets: Identification of glucose-responsive genes and a highly regulated tgfbeta signaling pathway. Endocrinology 2002, 143, 3695–3698. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces akt/bcl-xl signaling and pancreatic beta-cell mass and protects against diabetes. Faseb J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Koren, S.; Razik, F.; Goldberg, H.; Kwan, E.P.; Sheu, L.; Gaisano, H.Y.; Fantus, I.G. High beta-cell mass prevents streptozotocin-induced diabetes in thioredoxin-interacting protein-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1251–E1261. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Saxena, G.; Chen, J.; Shalev, A. Glucose-stimulated expression of txnip is mediated by carbohydrate response element-binding protein, p300, and histone h4 acetylation in pancreatic beta cells. J. Biol. Chem. 2009, 284, 16898–16905. [Google Scholar] [CrossRef] [PubMed]
- Decaux, J.F.; Antoine, B.; Kahn, A. Regulation of the expression of the l-type pyruvate kinase gene in adult rat hepatocytes in primary culture. J. Biol. Chem. 1989, 264, 11584–11590. [Google Scholar] [PubMed]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (chrebp) and sterol regulatory element binding protein-1c (srebp-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, C.; Chen, J.; Xu, G.; Jing, G.; Shalev, A. Foxo1 competes with carbohydrate response element-binding protein (chrebp) and inhibits thioredoxin-interacting protein (txnip) transcription in pancreatic beta cells. J. Biol. Chem. 2013, 288, 23194–23202. [Google Scholar] [CrossRef]
- Tsatsos, N.G.; Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Identification and function of phosphorylation in the glucose-regulated transcription factor chrebp. Biochem. J. 2008, 411, 261–270. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-glcnacylation increases chrebp protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of chrebp-dependent hepatic steatosis in mice. J. Clin. Invest. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel chrebp isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (chrebp). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Takeda, J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic beta cell line, ins-1e. Endocr. J. 2013, 60, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other chrebp target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef]
- Frodin, M.; Gammeltoft, S. Role and regulation of 90 kda ribosomal s6 kinase (rsk) in signal transduction. Mol. Cell Endocrinol. 1999, 151, 65–77. [Google Scholar] [CrossRef]
- Anjum, R.; Blenis, J. The rsk family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef]
- Abe, J.; Okuda, M.; Huang, Q.; Yoshizumi, M.; Berk, B.C. Reactive oxygen species activate p90 ribosomal s6 kinase via fyn and ras. J. Biol. Chem. 2000, 275, 1739–1748. [Google Scholar] [CrossRef]
- da Costa-Pessoa, J.M.; Damasceno, R.S.; Machado, U.F.; Beloto-Silva, O.; Oliveira-Souza, M. High glucose concentration stimulates nhe-1 activity in distal nephron cells: The role of the mek/erk1/2/p90rsk and p38mapk signaling pathways. Cell Physiol. Biochem. 2014, 33, 333–343. [Google Scholar] [CrossRef]
- Le, N.T.; Takei, Y.; Shishido, T.; Woo, C.H.; Chang, E.; Heo, K.S.; Lee, H.; Lu, Y.; Morrell, C.; Oikawa, M.; et al. P90rsk targets the erk5-chip ubiquitin e3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ. Res. 2012, 110, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Heo, K.S.; Takei, Y.; Lee, H.; Woo, C.H.; Chang, E.; McClain, C.; Hurley, C.; Wang, X.; Li, F.; et al. A crucial role for p90rsk-mediated reduction of erk5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 2013, 127, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ibanez, O.; Affo, S.; Rodrigo-Torres, D.; Blaya, D.; Millan, C.; Coll, M.; Perea, L.; Odena, G.; Knorpp, T.; Templin, M.F.; et al. Kinase analysis in alcoholic hepatitis identifies p90rsk as a potential mediator of liver fibrogenesis. Gut 2016, 65, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Ding, B.; Shishido, T.; Lerner-Marmarosh, N.; Wang, N.; Maekawa, N.; Berk, B.C.; Takeishi, Y.; Yan, C.; Blaxall, B.C.; et al. Role of p90 ribosomal s6 kinase-mediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation 2006, 113, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; White, S.A.; Hu, K. Role of p90rsk in kidney and other diseases. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Poteet-Smith, C.E.; Xu, Y.; Errington, T.M.; Hecht, S.M.; Lannigan, D.A. Identification of the first specific inhibitor of p90 ribosomal s6 kinase (rsk) reveals an unexpected role for rsk in cancer cell proliferation. Cancer Res. 2005, 65, 1027–1034. [Google Scholar] [PubMed]
- Sapkota, G.P.; Cummings, L.; Newell, F.S.; Armstrong, C.; Bain, J.; Frodin, M.; Grauert, M.; Hoffmann, M.; Schnapp, G.; Steegmaier, M.; et al. Bi-d1870 is a specific inhibitor of the p90 rsk (ribosomal s6 kinase) isoforms in vitro and in vivo. Biochem. J. 2007, 401, 29–38. [Google Scholar] [CrossRef]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef]
- Efanova, I.B.; Zaitsev, S.V.; Zhivotovsky, B.; Kohler, M.; Efendic, S.; Orrenius, S.; Berggren, P.O. Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular ca2+ concentration. J. Biol. Chem. 1998, 273, 33501–33507. [Google Scholar] [CrossRef]
- Roche, E.; Farfari, S.; Witters, L.A.; Assimacopoulos-Jeannet, F.; Thumelin, S.; Brun, T.; Corkey, B.E.; Saha, A.K.; Prentki, M. Long-term exposure of beta-ins cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 1998, 47, 1086–1094. [Google Scholar] [CrossRef]
- Kasuga, M. Insulin resistance and pancreatic beta cell failure. J. Clin. Invest. 2006, 116, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Rachdi, L.; Weiss, A.J.; Cras-Meneur, C.; Bernal-Mizrachi, E. Regulation of beta-cell mass and function by the akt/protein kinase b signalling pathway. Diabetes Obes. Metab. 2007, 9 (Suppl. 2), 147–157. [Google Scholar] [CrossRef]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. Mafa is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed]
- Salakou, S.; Kardamakis, D.; Tsamandas, A.C.; Zolota, V.; Apostolakis, E.; Tzelepi, V.; Papathanasopoulos, P.; Bonikos, D.S.; Papapetropoulos, T.; Petsas, T.; et al. Increased bax/bcl-2 ratio up-regulates caspase-3 and increases apoptosis in the thymus of patients with myasthenia gravis. In Vivo 2007, 21, 123–132. [Google Scholar] [PubMed]
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Wieder, T.; Sturm, I.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. The bax/bcl-2 ratio determines the susceptibility of human melanoma cells to cd95/fas-mediated apoptosis. J. Investig. Dermatol. 2001, 117, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates er stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Hadjivassiliou, H.; Taunton, J. A clickable inhibitor reveals context-dependent autoactivation of p90 rsk. Nat. Chem. Biol. 2007, 3, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by mondoa:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet chrebp-beta is increased in diabetes and controls chrebp-alpha and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (chrebp) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Robinson, L.N.; Towle, H.C. Chrebp*mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferre, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of chrebp in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P.E.; Kostianovsky, M. Method for the isolation of intact islets of langerhans from the rat pancreas. Diabetes 1967, 16, 35–39. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.-H.; Kim, S.; Kim, S.; Lee, H.; Park, S.-Y.; Woo, C.-H. FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic ? Cells. Int. J. Mol. Sci. 2019, 20, 4424. https://doi.org/10.3390/ijms20184424
Han J-H, Kim S, Kim S, Lee H, Park S-Y, Woo C-H. FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic ? Cells. International Journal of Molecular Sciences. 2019; 20(18):4424. https://doi.org/10.3390/ijms20184424
Chicago/Turabian StyleHan, Jung-Hwa, Suji Kim, Sujin Kim, Heejung Lee, So-Young Park, and Chang-Hoon Woo. 2019. "FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic ? Cells" International Journal of Molecular Sciences 20, no. 18: 4424. https://doi.org/10.3390/ijms20184424
APA StyleHan, J.-H., Kim, S., Kim, S., Lee, H., Park, S.-Y., & Woo, C.-H. (2019). FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic ? Cells. International Journal of Molecular Sciences, 20(18), 4424. https://doi.org/10.3390/ijms20184424