Different Synergy in Amyloids and Biologically Active Forms of Proteins

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Comparative Analysis of the ASyn Structure
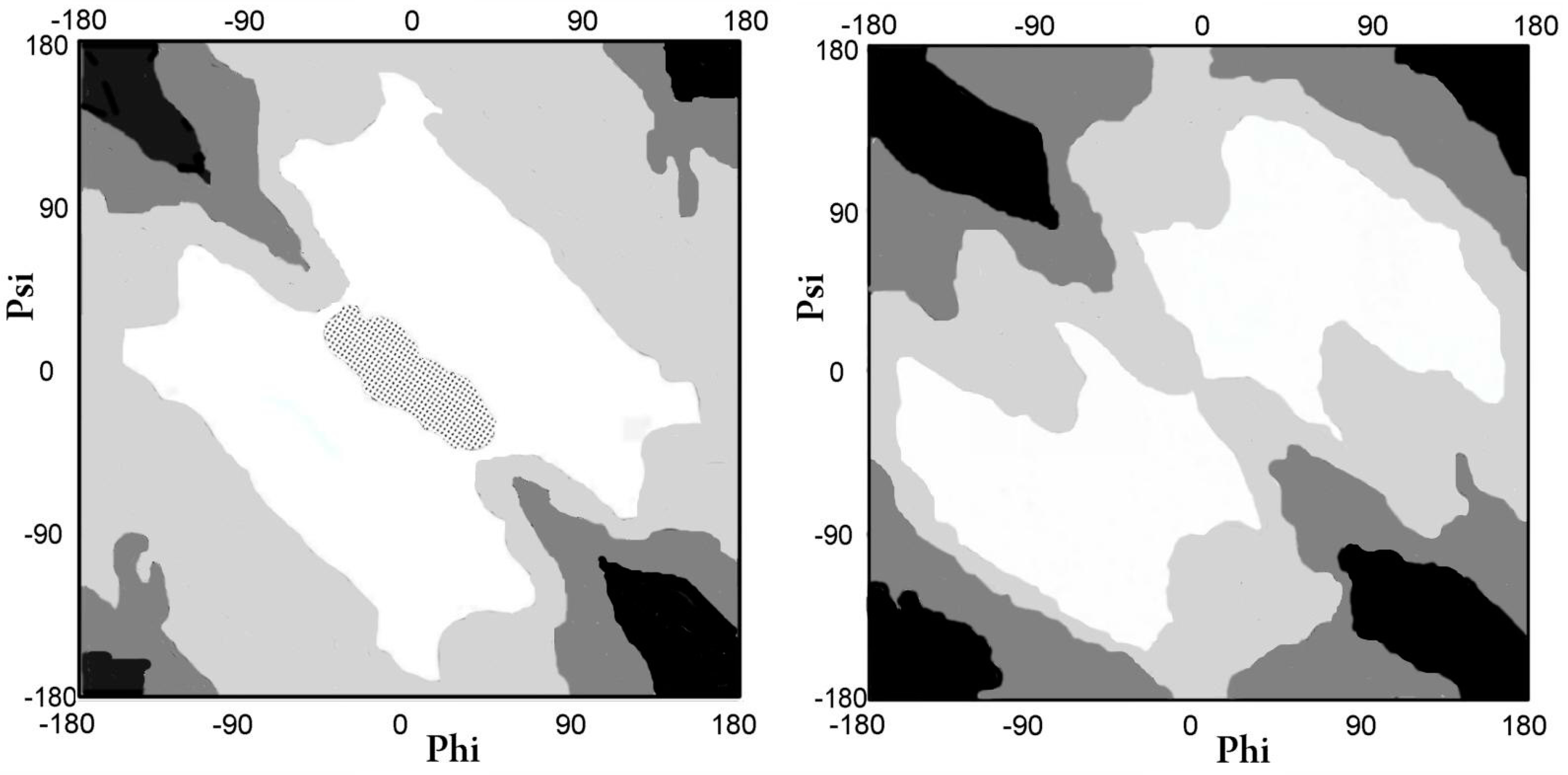
2.1.1. Comparative Analysis of the Distribution of Phi and Psi Angles of the Asyn Structure in the Form of Micelle-bound (Single Molecule) and in the Form of Amyloid Fibril
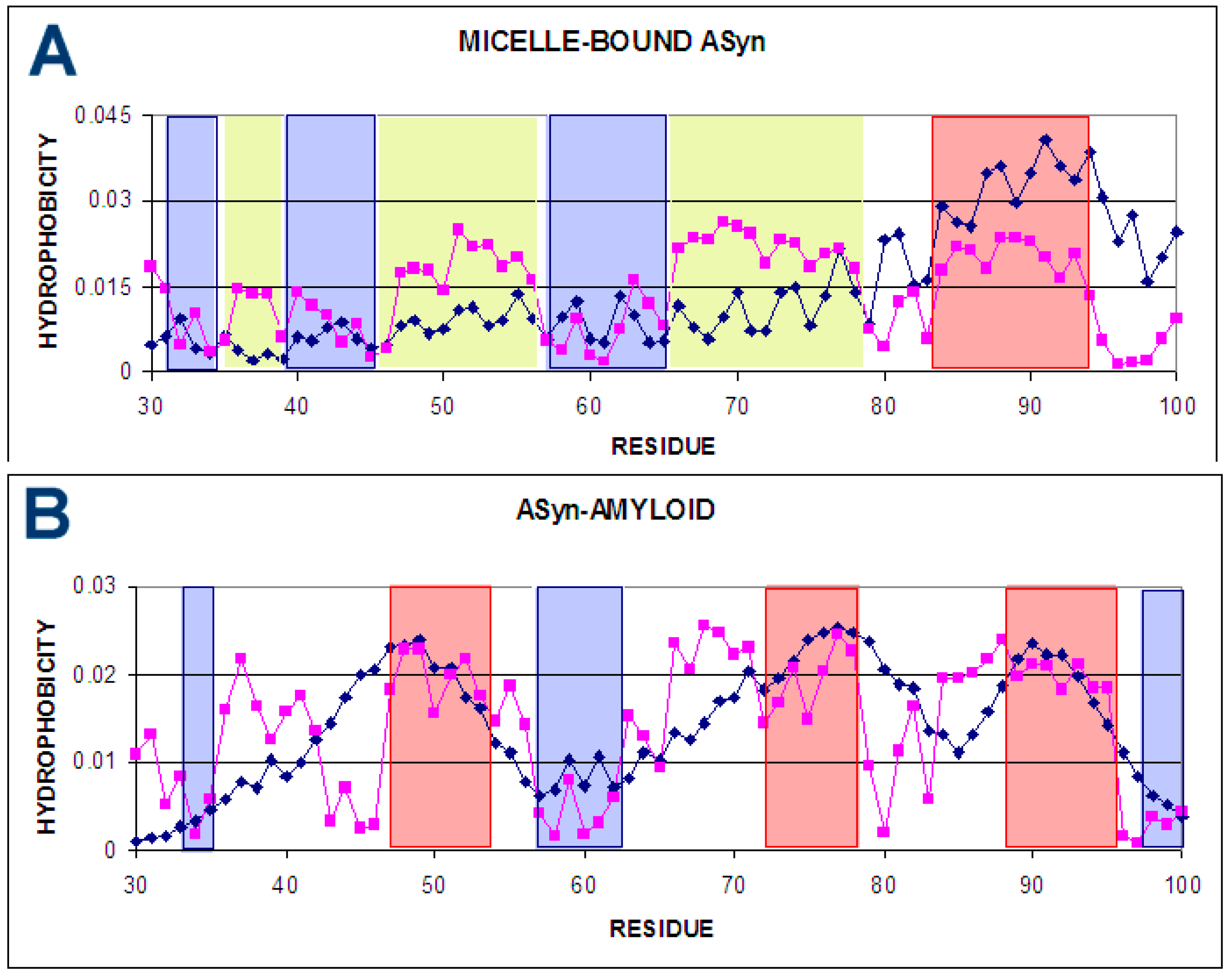
2.1.2. Determining the Status of Individual Residues in Relation to the Structure of the Hydrophobic Core
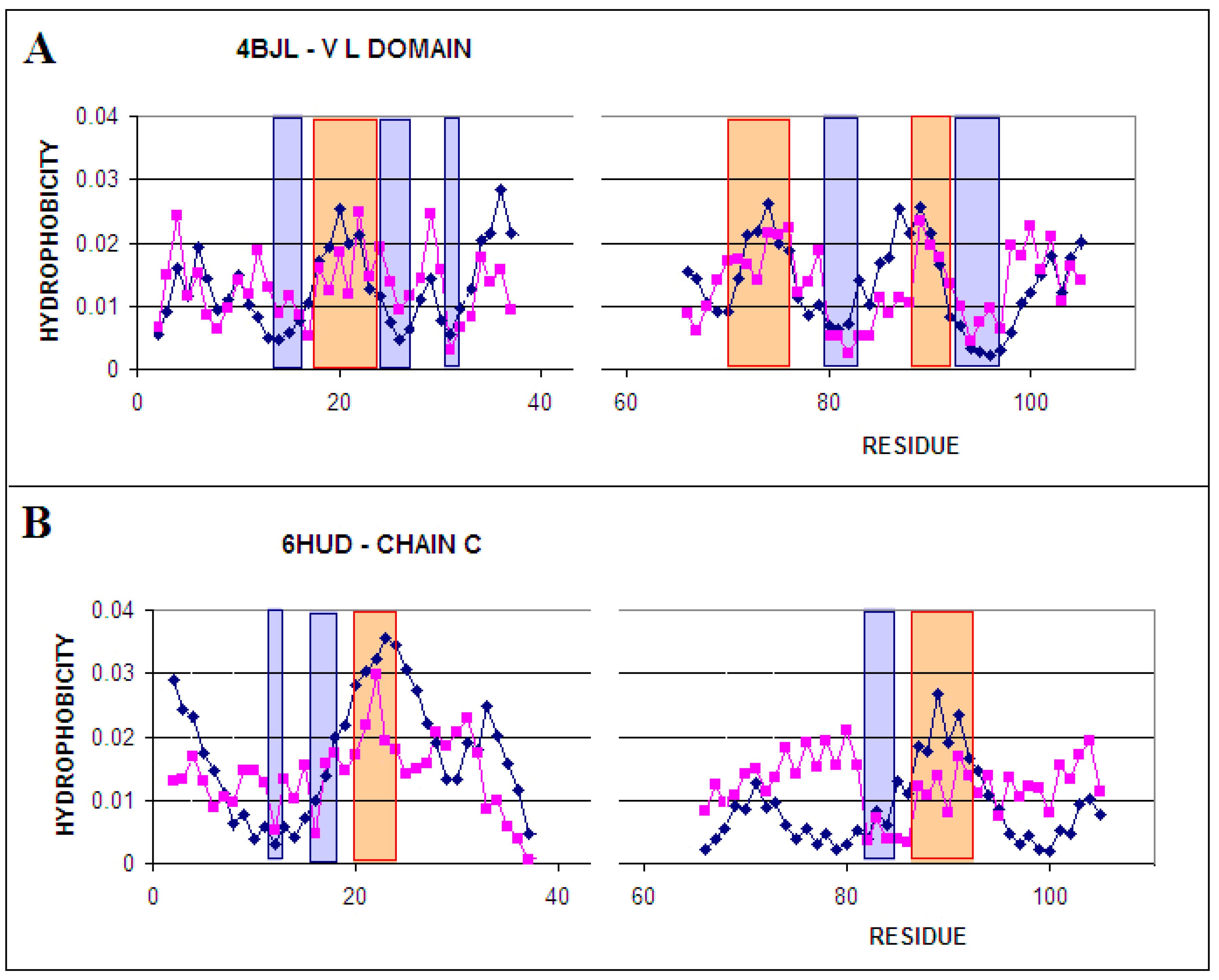
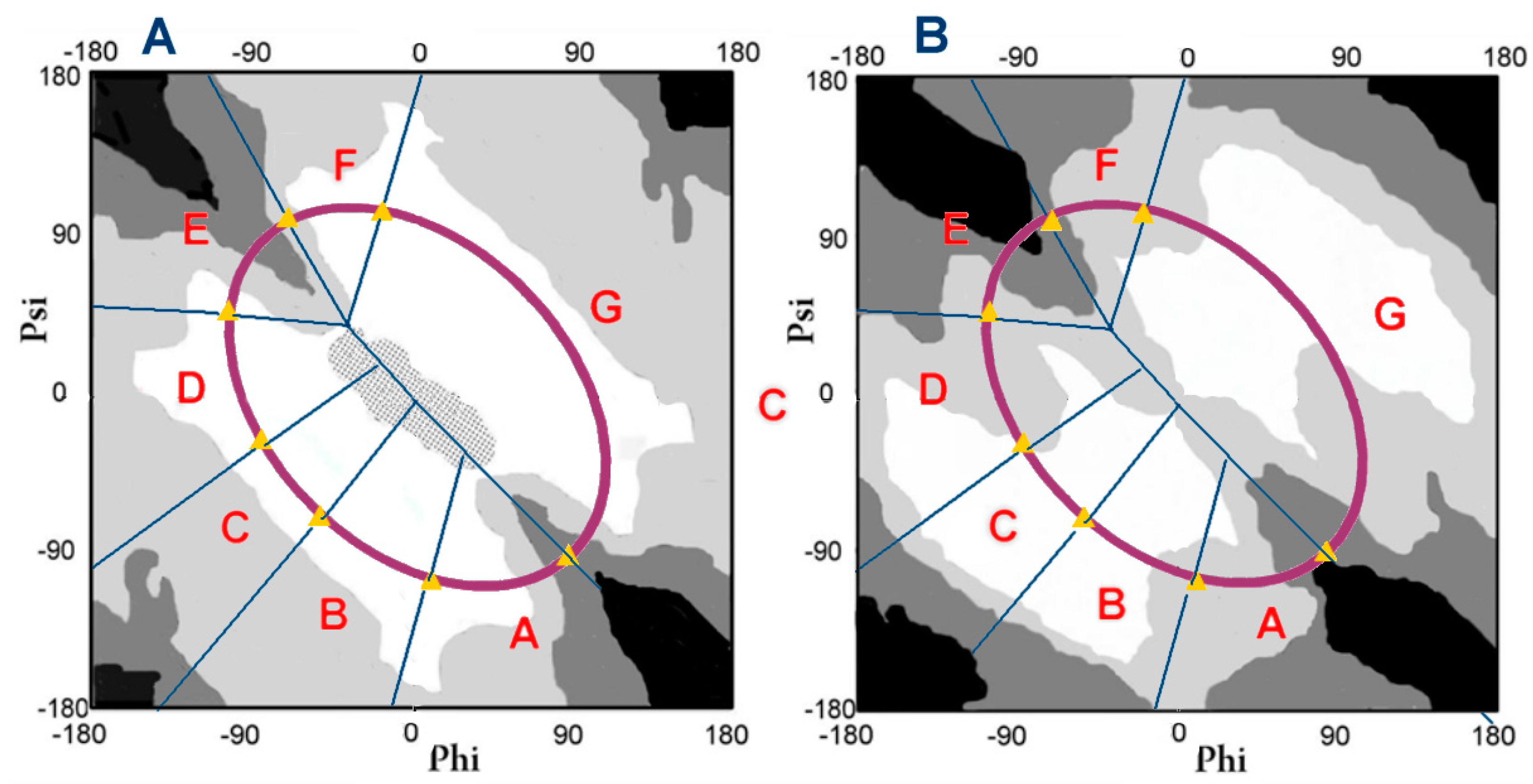
2.2. Analysis of the V Domain of the Light Chain (lambda) of Immunoglobulin G in its Amyloid Form
2.2.1. Comparative Analysis of Phi and Psi Angle Distribution of the V Domain of Immunoglobulin G in Native Form (Bence–Jones Protein—Light Chain Dimer) and in the Form of Amyloid Fibril
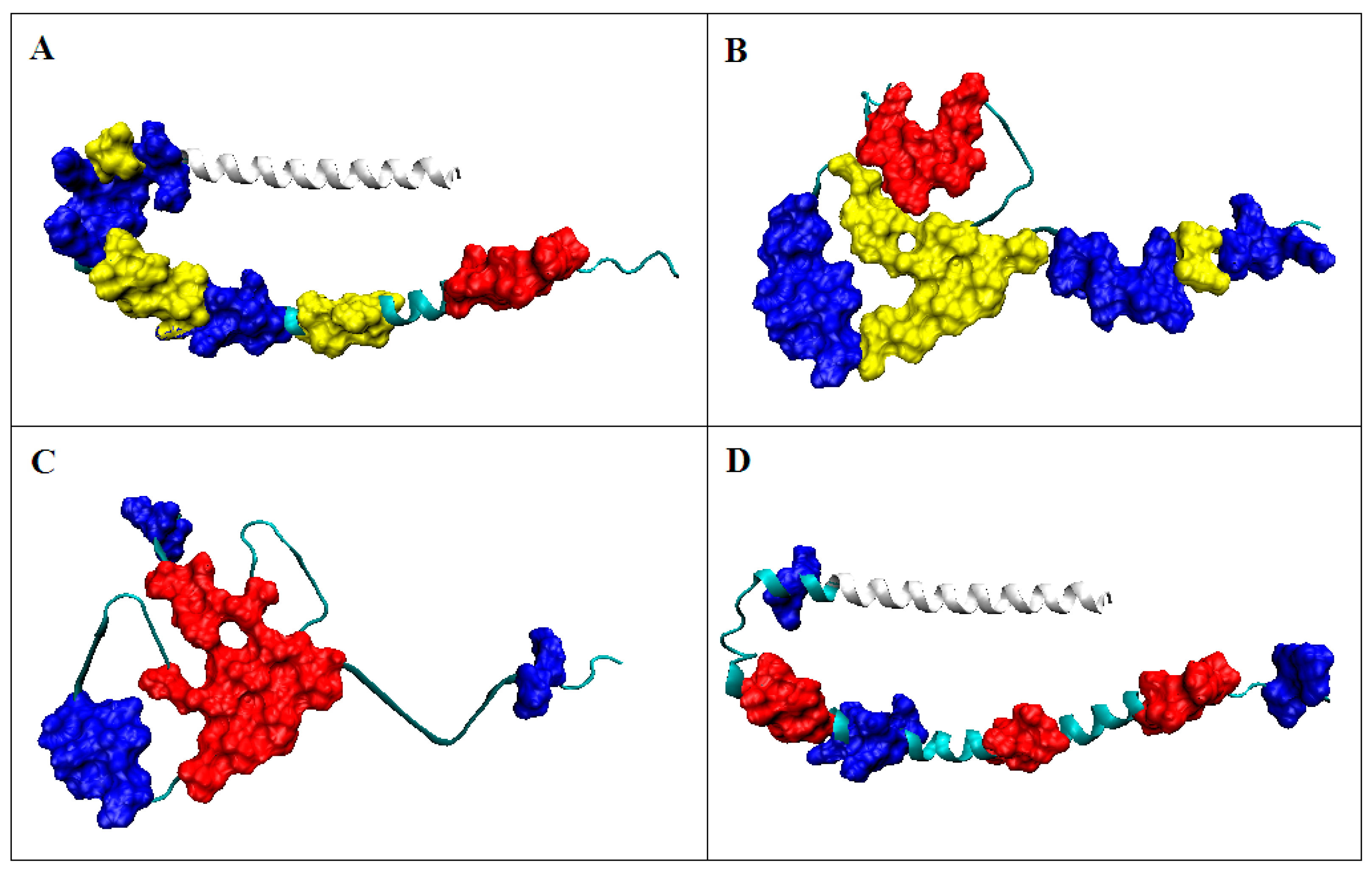
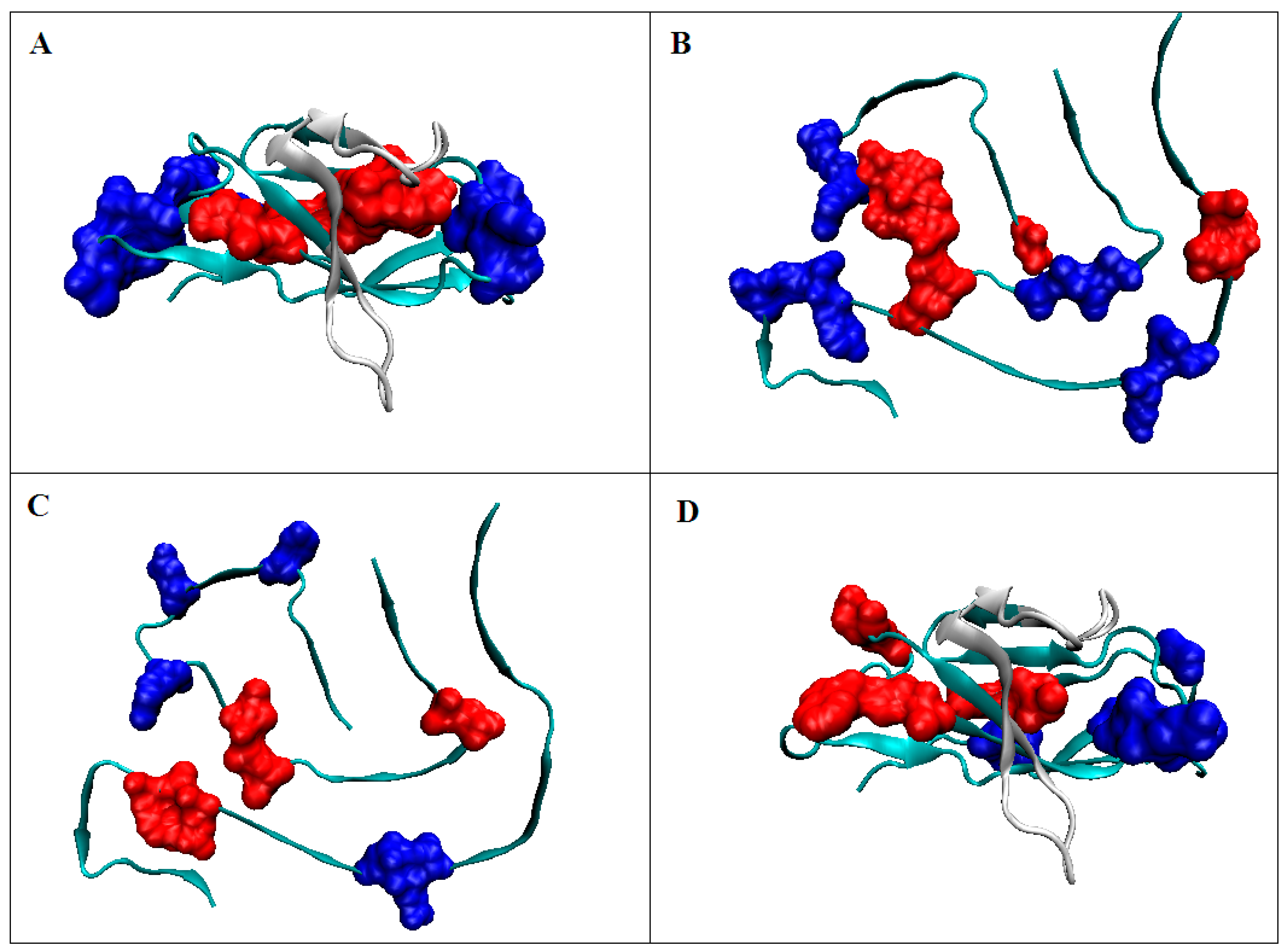
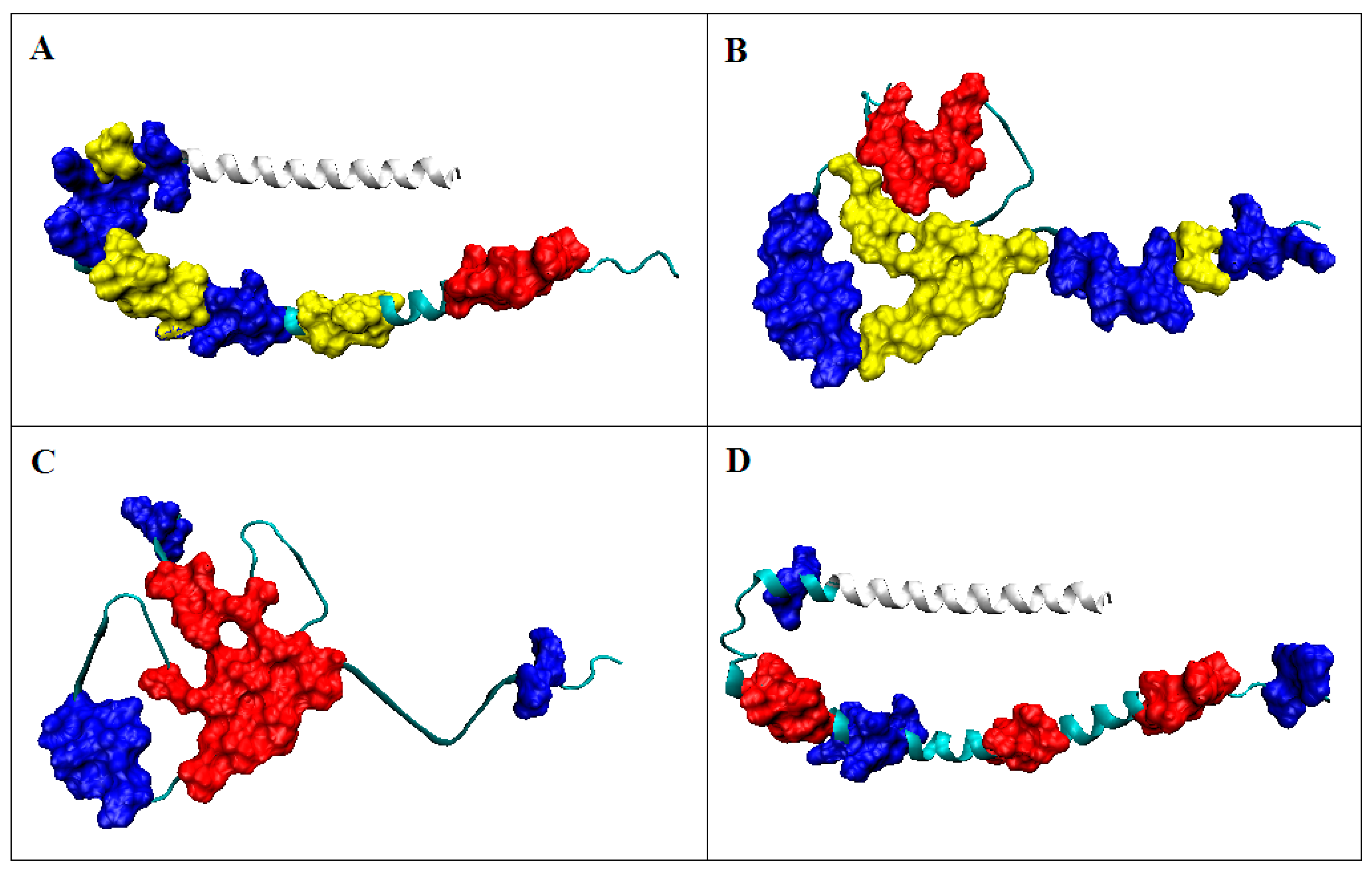
2.2.2. Participation of the V domain in the Construction of Complexes in the VL-VL (Bence–Jones Dimer) and VL-VH (FAB Fragment of IgG) Fragments
3. Materials and Methods
3.1. Structural Codes of the Polypeptide Chain Form—Geometric Interpretation
3.2. Data
3.3. Calculation Procedure
3.4. Protein Status Analysis Taking into Account the Synergy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASyn | α-synuclein |
References
- Dill, K.A.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Ann. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell. Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. The “Correctly Folded” state of proteins, is it a metastable state? Angew. Chem. Int. Ed. Engl. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Dobson, C.M.; Šali, A.; Karplus, M. Protein Folding, A Perspective from Theory and Experiment. Angew. Chem. Int. Ed Engl. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowlesm, T.P.J.; Tartagliam, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Tycko, R. Alzheimer’s disease, Structure of aggregates revealed. Nature 2016, 537, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Molecular Structure of Aggregated Amyloid-β, Insights from Solid-State Nuclear Magnetic Resonance. Cold Spring Harb. Perspect. Med. 2016, 6, a024083. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yau, W.M.; Qiang, W. Modeling an in-register, parallel “iowa” Aβ fibril structure using solid-state NMR data from labeled samples with rosetta. Structure 2015, 623, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed. Engl. 2015, 54, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef]
- Huang, D.B.; Ainsworth, C.F.; Stevens, F.J.; Schiffer, M. Three quaternary structures for a single protein. Proc. Natl. Acad. Sci. USA 1996, 93, 7017–7021. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, B.; Banach, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Is the hydrophobic core a universal structural element in proteins? J. Mol. Model. 2017, 23, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. Secondary and Supersecondary Structure of Proteins in Light of the Structure of Hydrophobic Cores. Methods Mol. Biol. 2019, 1958, 347–378. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Banach, M.; Konieczny, L. Application of the Fuzzy Oil Drop Model Describes Amyloid as a Ribbonlike Micelle. Entropy 2017, 19, 167. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Kalinowska, B.; Alejster, P.; Sałapa, K.; Baster, Z.; Roterman, I. Hypothetical in silico model of the early-stage intermediate in protein folding. J. Mol. Model. 2013, 19, 4259–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowska, B.; Fabian, P.; Stąpor, K.; Roterman, I. Statistical dictionaries for hypothetical in silico model of the early-stage intermediate in protein folding. J. Comput. Aided Mol. Des. 2015, 29, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roterman, I. The geometrical analysis of peptide backbone structure and its local deformations. Biochimie 1995, 77, 204–216. [Google Scholar] [CrossRef]
- Roterman, I. Modelling the optimal simulation path in the peptide chain folding—studies based on geometry of alanine heptapeptide. J. Biol. 1995, 177, 283–288. [Google Scholar] [CrossRef]
- Dułak, D.; Banach, M.; Gadzała, M.; Konieczny, L.; Roterman, I. Structural analysis of the Aβ(15-40) amyloid fibril based on hydrophobicity distribution. Acta Biochim. Pol. 2018, 65, 595–604. [Google Scholar] [CrossRef]
- Roterman, I.; Dułak, D.; Gadzała, M.; Banach, M.; Konieczny, L. Structural analysis of the Aβ(11-42) amyloid fibril based on hydrophobicity distribution. J. Comput. Aided Mol. Des. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dułak, D.; Gadzała, M.; Banach, M.; Ptak, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Filamentous Aggregates of Tau Proteins Fulfil Standard Amyloid Criteria Provided by the Fuzzy Oil Drop (FOD) Model. Int. J. Mol. Sci. 2018, 19, 2910. [Google Scholar] [CrossRef] [PubMed]
- Harry, W.; Schroeder, H.W., Jr.; Cavacini, L. Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef]
- Kalinowska, B.; Krzykalski, A.; Roterman, I. Contingency Table Browser—Prediction of early stage protein structure. Bioinformation 2015, 11, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Konieczny, L.; Jurkowski, W.; Prymula, K.; Banach, M. Two-intermediate model to characterize the structure of fast-folding proteins. J. Biol. 2011, 283, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef]
- Konieczny, L.; Bryliński, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Biol. 2006, 6, 15–22. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Jurkowski, W.; Baster, Z.; Dułak, D.; Roterman, I. The early-stage intermediate. In Protein Folding in Silico Ed. Irena Roterman-Konieczna; Woodhead Publishing (currently Elsevier): Oxford, UK; Cambridge, UK; Philadelphia, PA, USA; New Dehli, India, 2012; pp. 1–20. [Google Scholar]
- Fabian, P.; Stapor, K.; Roterman, I. Model of early stage intermediate in respect to its final structure. Biochimie 2019. submitted. [Google Scholar]
- Alejster, P.; Jurkowski, W.; Roterman, I. Structural information involved in the interpretation of the stepwise protein folding process. In Protein Folding in Silico Ed. Irena Roterman-Konieczna; Woodhead Publishing (currently Elsevier): Oxford, UK, 2012; pp. 39–54. [Google Scholar]
- Vogan, E.M.; Bellamacina, C.; He, X.; Liu, H.W.; Ringe, D.; Petsko, G.A. Crystal structure at 1.8 A resolution of CDP-D-glucose 4,6-dehydratase from Yersinia pseudotuberculosis. Biochemie 2004, 43, 3057–3067. [Google Scholar] [CrossRef]
- Available online: http, //www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html (accessed on 10 August 2019).
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum, Structural summaries of PDB entries. Prot. Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]


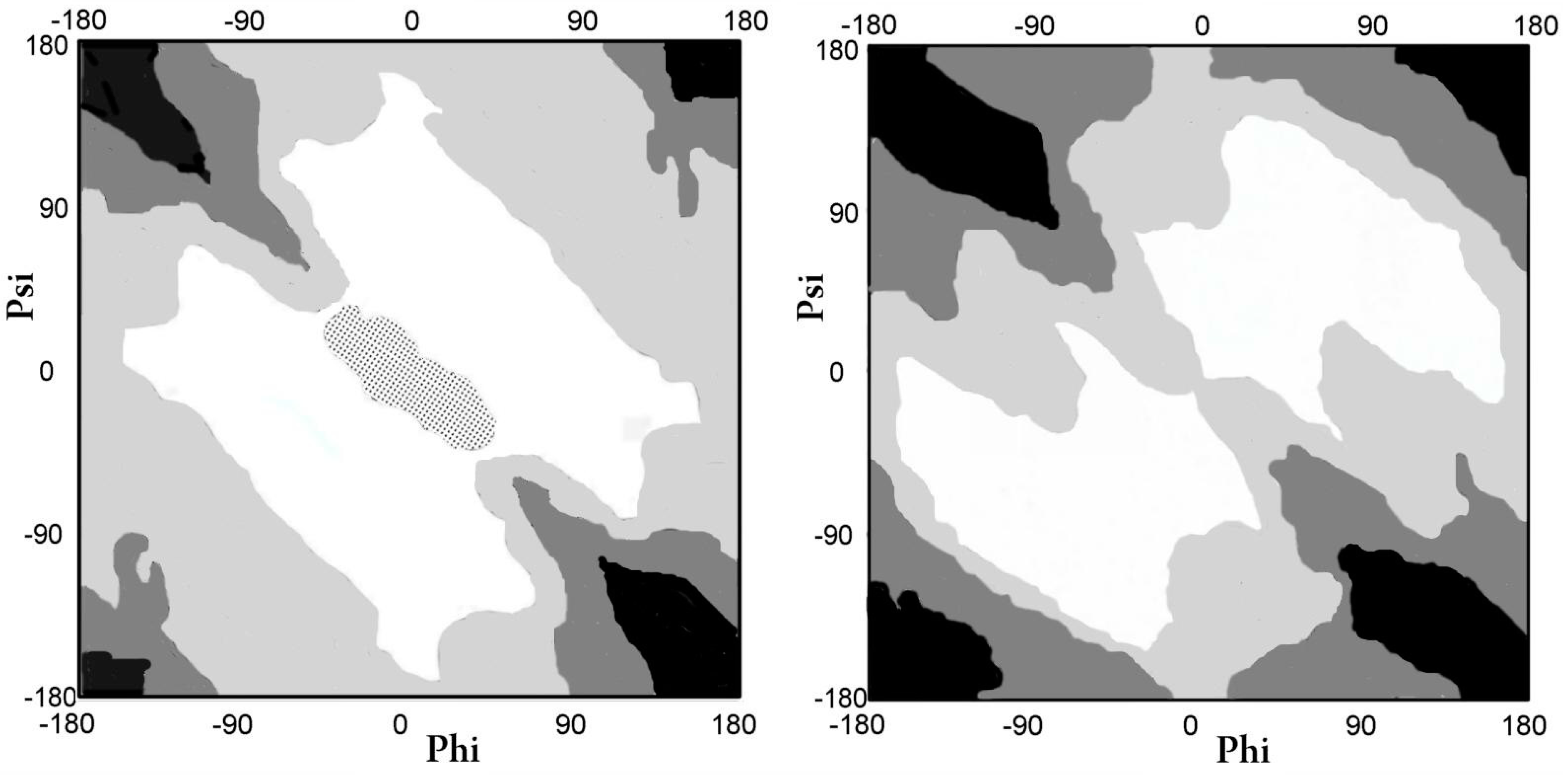

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1XQ8 code → 2N0A code ↓ | A | B | C | D | E | F | G | Σ |
|---|---|---|---|---|---|---|---|---|
| A | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| B | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 3 |
| C | 0 | 0 | 12 | 0 | 2 | 1 | 1 | 16 |
| D | 1 | 0 | 3 | 2 | 2 | 0 | 1 | 9 |
| E | 1 | 1 | 43 | 3 | 5 | 6 | 1 | 60 |
| F | 1 | 1 | 19 | 4 | 1 | 2 | 3 | 31 |
| G | 0 | 0 | 11 | 3 | 3 | 2 | 1 | 20 |
| Σ | 3 | 2 | 91 | 13 | 13 | 11 | 7 | 140 |
| ASyn → ASyn-FIBRIL ↓ | Hydrophobic Core | Status Accordant | Status Not-accordant | Σ |
|---|---|---|---|---|
| Hydrophobic Core | 4 | 2 | 19 | 25 |
| Accordant | 2 | 12 | 0 | 14 |
| Not-accordant | 5 | 15 | 12 | 32 |
| Σ | 11 | 29 | 31 | 71 |
| ASyn → Amyl Chain E ↓ | Hydrophobic Core | Accordant | Not-accordant | Σ |
|---|---|---|---|---|
| Hydr core | 11 | 2 | 15 | 28 |
| Accordant | 0 | 13 | 6 | 19 |
| Not-accordant | 0 | 13 | 11 | 24 |
| Σ | 11 | 28 | 32 | 71 |
| Chain E—in Fibril → Chain E—Individual ↓ | Hydrophobic Core | Accordant | Not-accordant | Σ |
|---|---|---|---|---|
| Hydr core | 16 | 3 | 9 | 28 |
| Accordant | 5 | 11 | 3 | 19 |
| Not-accordant | 4 | 0 | 20 | 24 |
| Σ | 25 | 14 | 32 | 71 |
| V domain → Fibril ↓ | B | C | D | E | F | G | Σ |
|---|---|---|---|---|---|---|---|
| A | 1 | 1 | |||||
| B | 1 | 1 | 1 | 3 | |||
| C | 1 | 1 | 1 | 1 | 4 | ||
| D | 0 | ||||||
| E | 2 | 6 (1) | 7 (3) | 28 (3) | 9 (2) | 5 | 57 (9) |
| F | 2 | 1 | 1 | 4 | |||
| G | 2 | 2 | |||||
| Σ | 3 | 6 (1) | 9 (3) | 32 (3) | 12 (2) | 9 | 71 (9) |
| Protein | Fragment | RD |
|---|---|---|
| V domain | 2–111 | 0.573 |
| (2–30) + (66–105) | 0.563 | |
| Fibril | ||
| Chains A–E | (1–30) + (66–105) | 0.773 |
| Chain C | (1–30) + (66–105) | 0.755 |
| V domain → Fibril ↓ | Hydrophobic Core | Accordant | Not-Accord | Σ |
|---|---|---|---|---|
| Hydrophobic Core | 6 (1) | 3 | 5 (2) | 14 (3) |
| Accordant | 7 (1) | 16 (3) | 1 | 24 (4) |
| Not Accordant | 8 (1) | 17 (1) | 9 | 34 (2) |
| Σ | 21 (3) | 36 (4) | 15 (2) | 72 (9) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabian, P.; Stapor, K.; Banach, M.; Ptak-Kaczor, M.; Konieczny, L.; Roterman, I. Different Synergy in Amyloids and Biologically Active Forms of Proteins. Int. J. Mol. Sci. 2019, 20, 4436. https://doi.org/10.3390/ijms20184436
Fabian P, Stapor K, Banach M, Ptak-Kaczor M, Konieczny L, Roterman I. Different Synergy in Amyloids and Biologically Active Forms of Proteins. International Journal of Molecular Sciences. 2019; 20(18):4436. https://doi.org/10.3390/ijms20184436
Chicago/Turabian StyleFabian, Piotr, Katarzyna Stapor, Mateusz Banach, Magdalena Ptak-Kaczor, Leszek Konieczny, and Irena Roterman. 2019. "Different Synergy in Amyloids and Biologically Active Forms of Proteins" International Journal of Molecular Sciences 20, no. 18: 4436. https://doi.org/10.3390/ijms20184436