Eicosapentaenoic Acid (EPA) Modulates Glucose Metabolism by Targeting AMP-Activated Protein Kinase (AMPK) Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
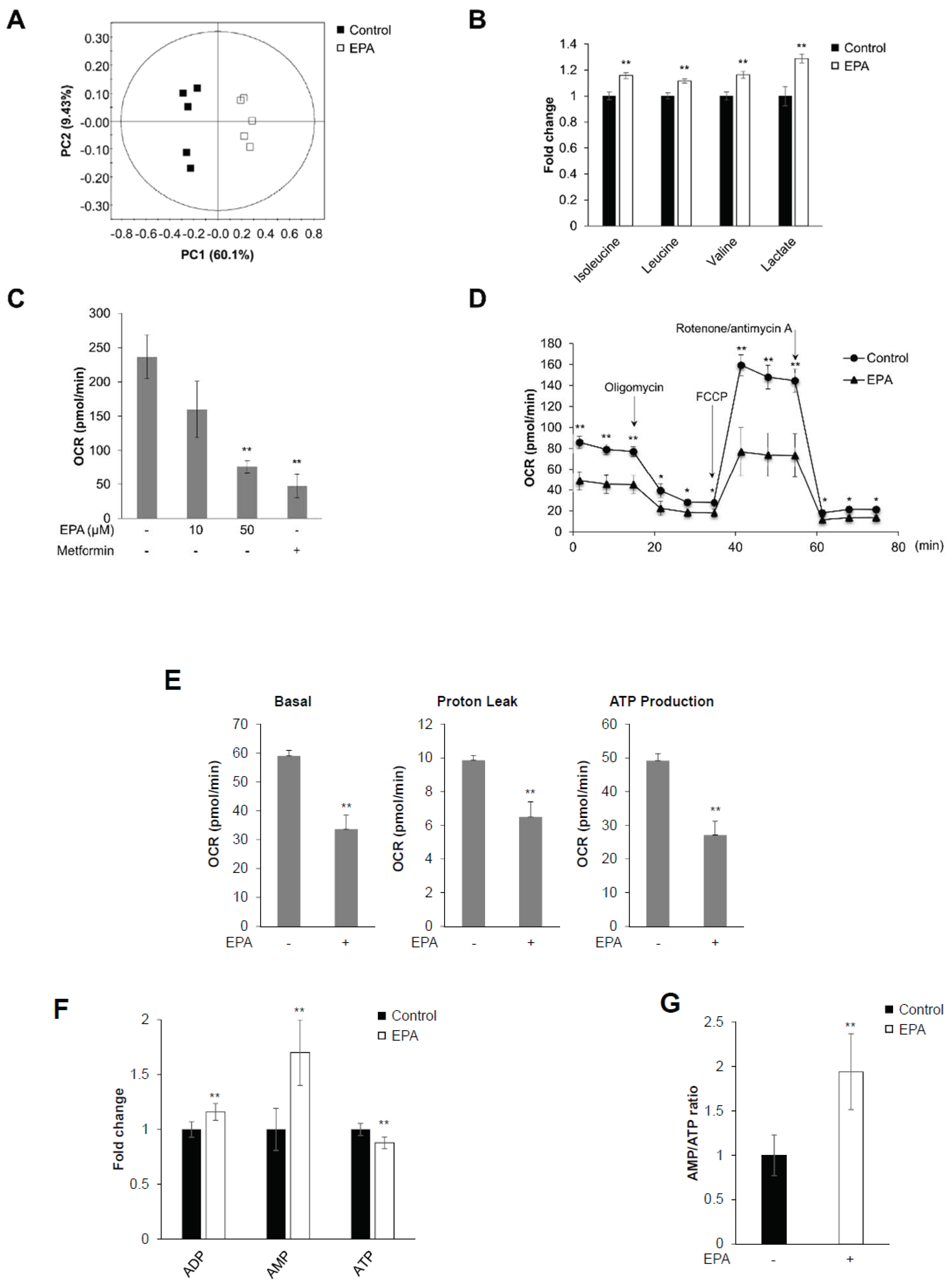
2.1. EPA Inhibits Mitochondrial Oxygen Consumption Rate (OCR) and Decreases Intracellular AMP:ATP Ratio in Skeletal Muscle Cells
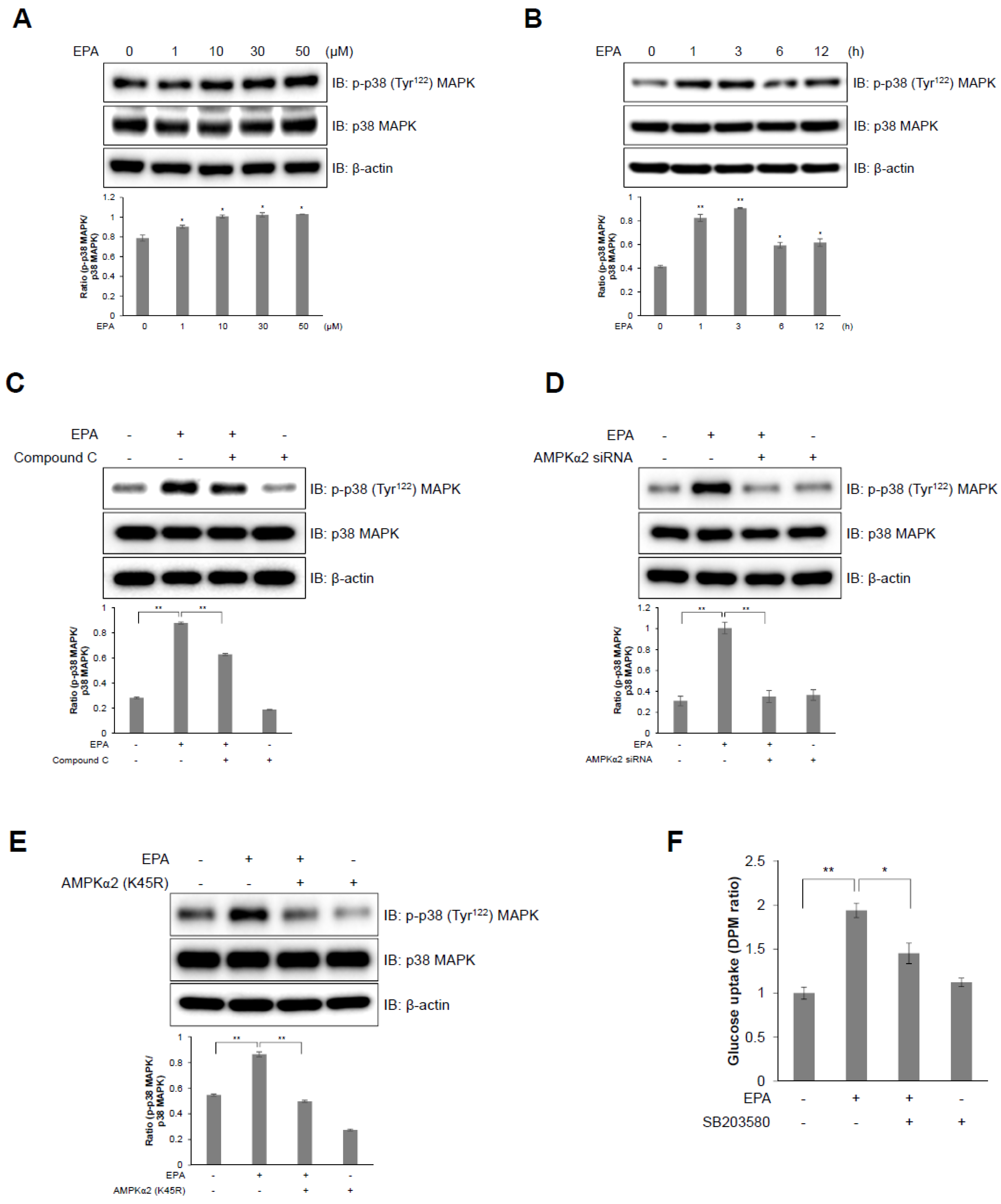
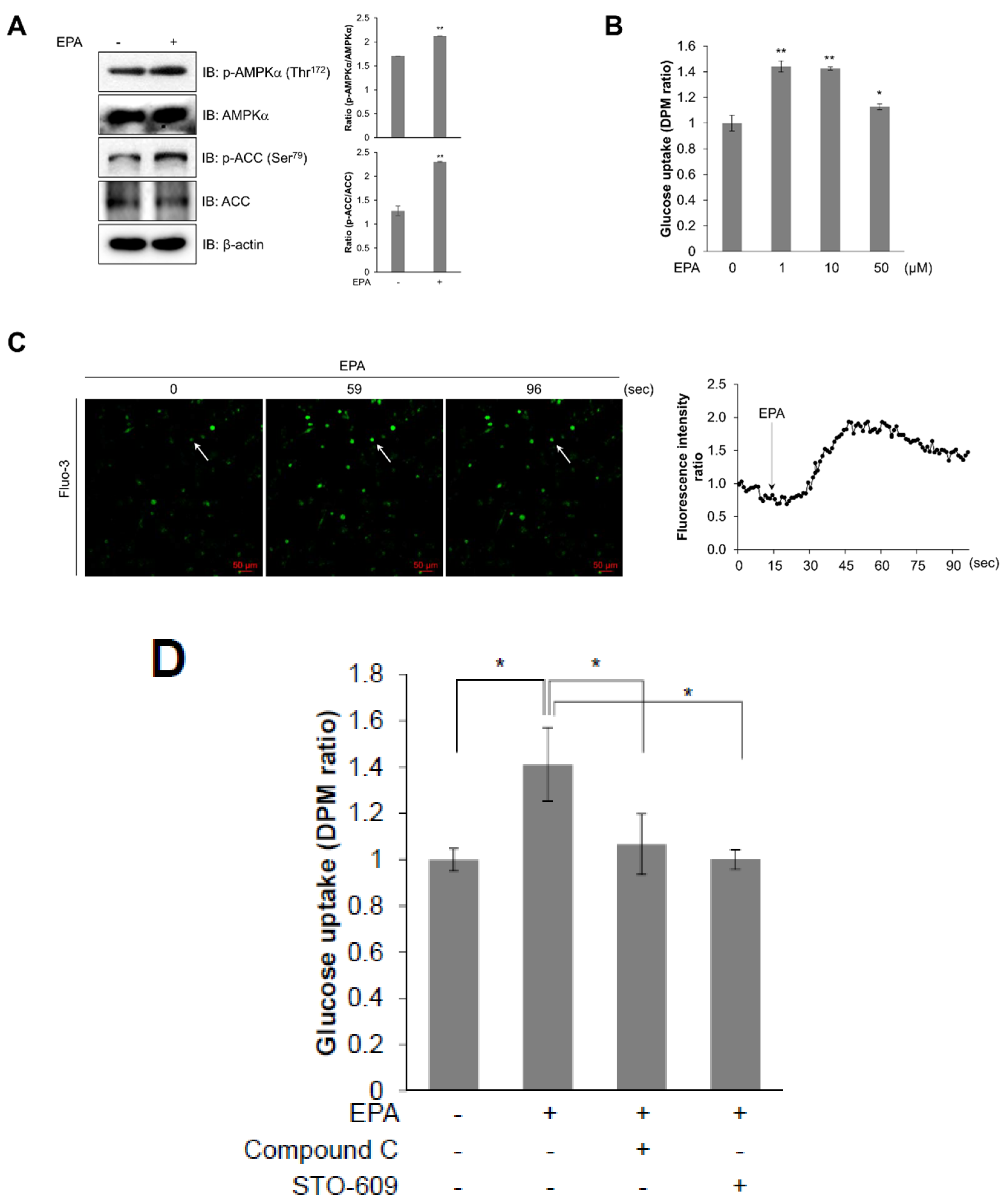
2.2. EPA Stimulates Glucose Uptake through the AMPK Signaling Pathway in C2C12 Skeletal Muscle Cells
2.3. Intracellular Calcium Plays an Upstream Role of AMPK in EPA-Mediated Glucose Uptake in Skeletal Muscle Cells
2.4. I p38 MAPK Plays a Downstream Role of AMPK in EPA-Mediated Glucose Uptake in C2C12 Skeletal Muscle Cells
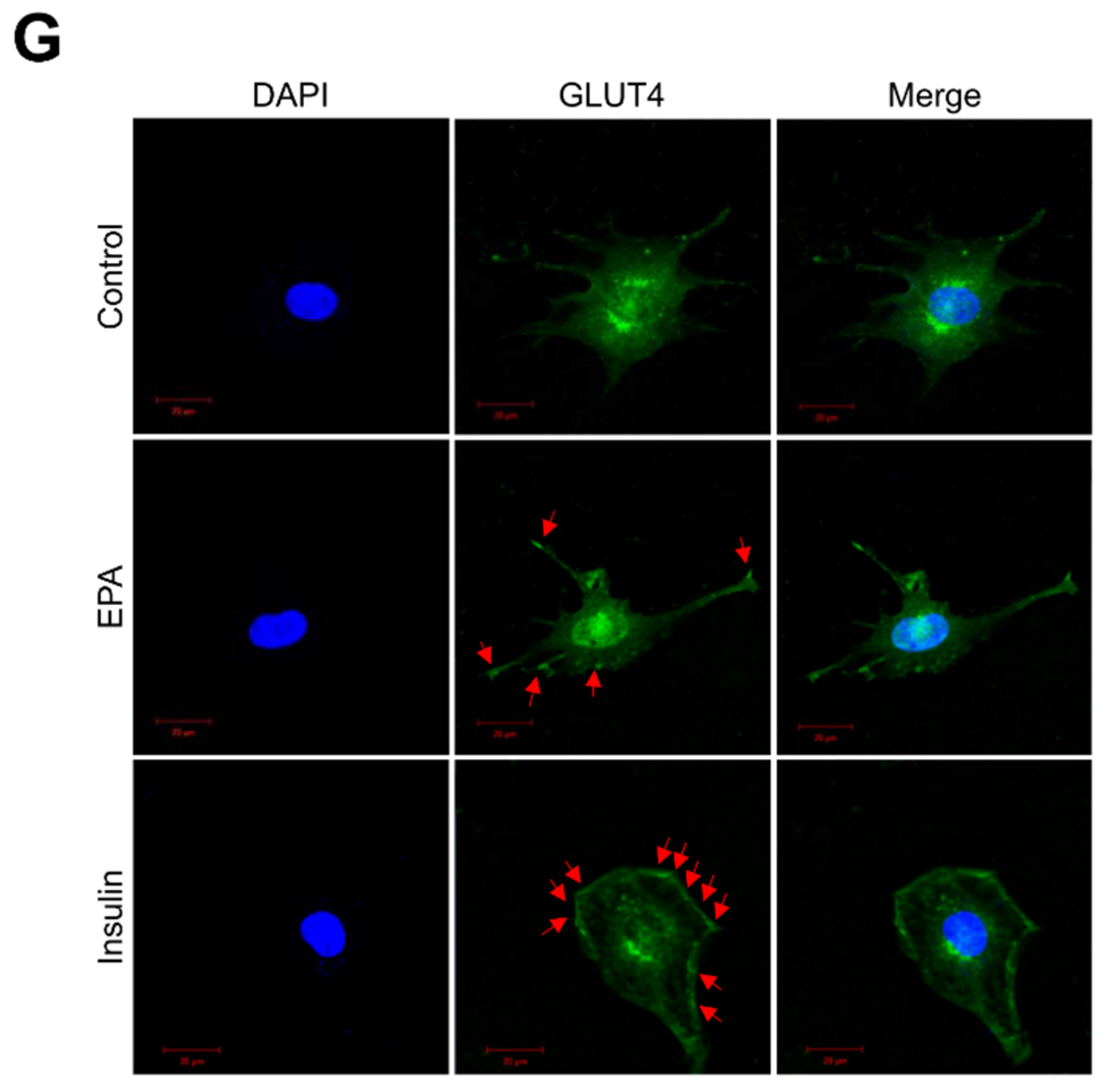
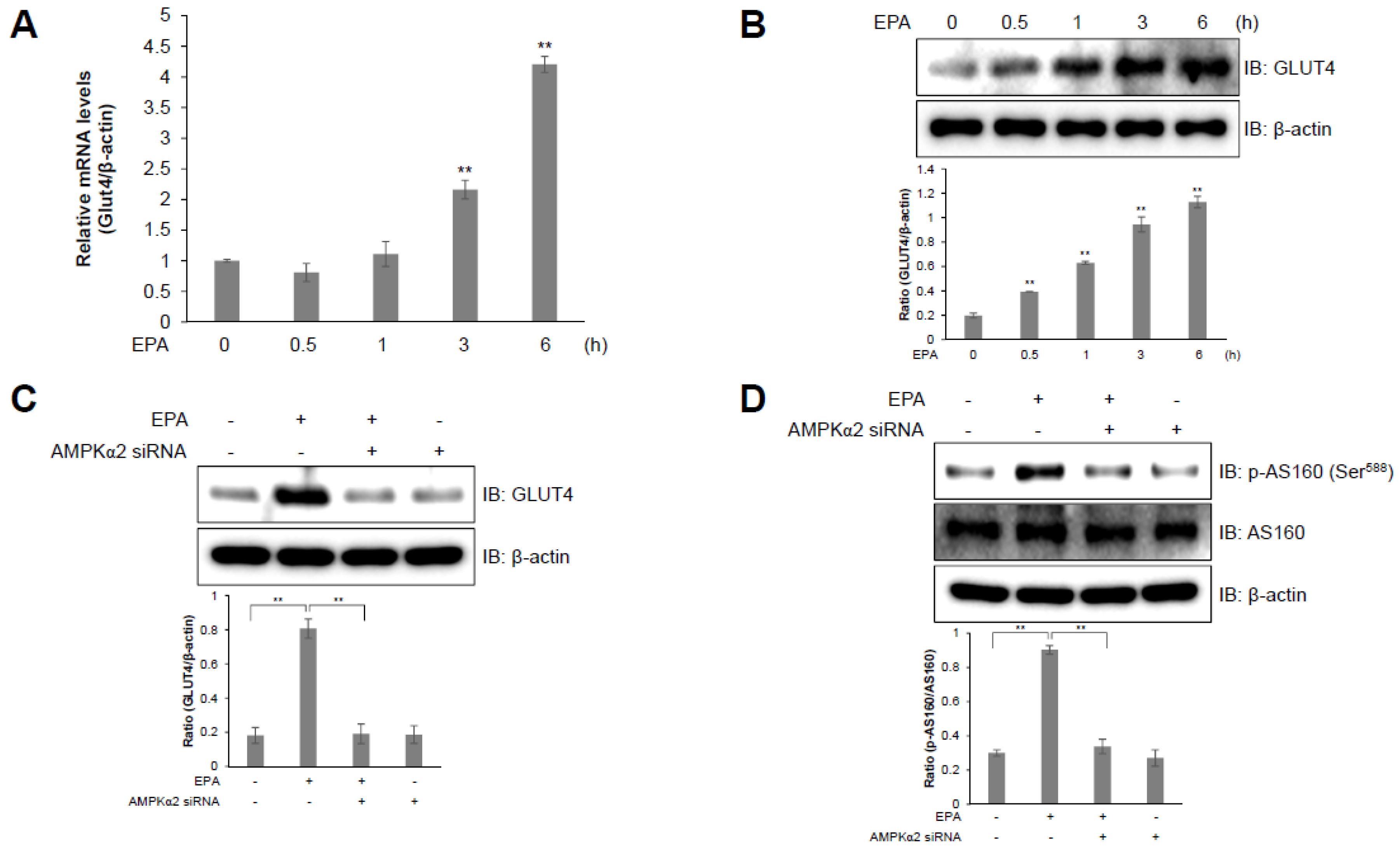
2.5. AS160 is Involved in EPA-Induced GLUT4 Expression in C2C12 Skeletal Muscle Cells
2.6. EPA Activates AMPK and Stimulates Glucose Uptake in Primary Cultured Myoblasts
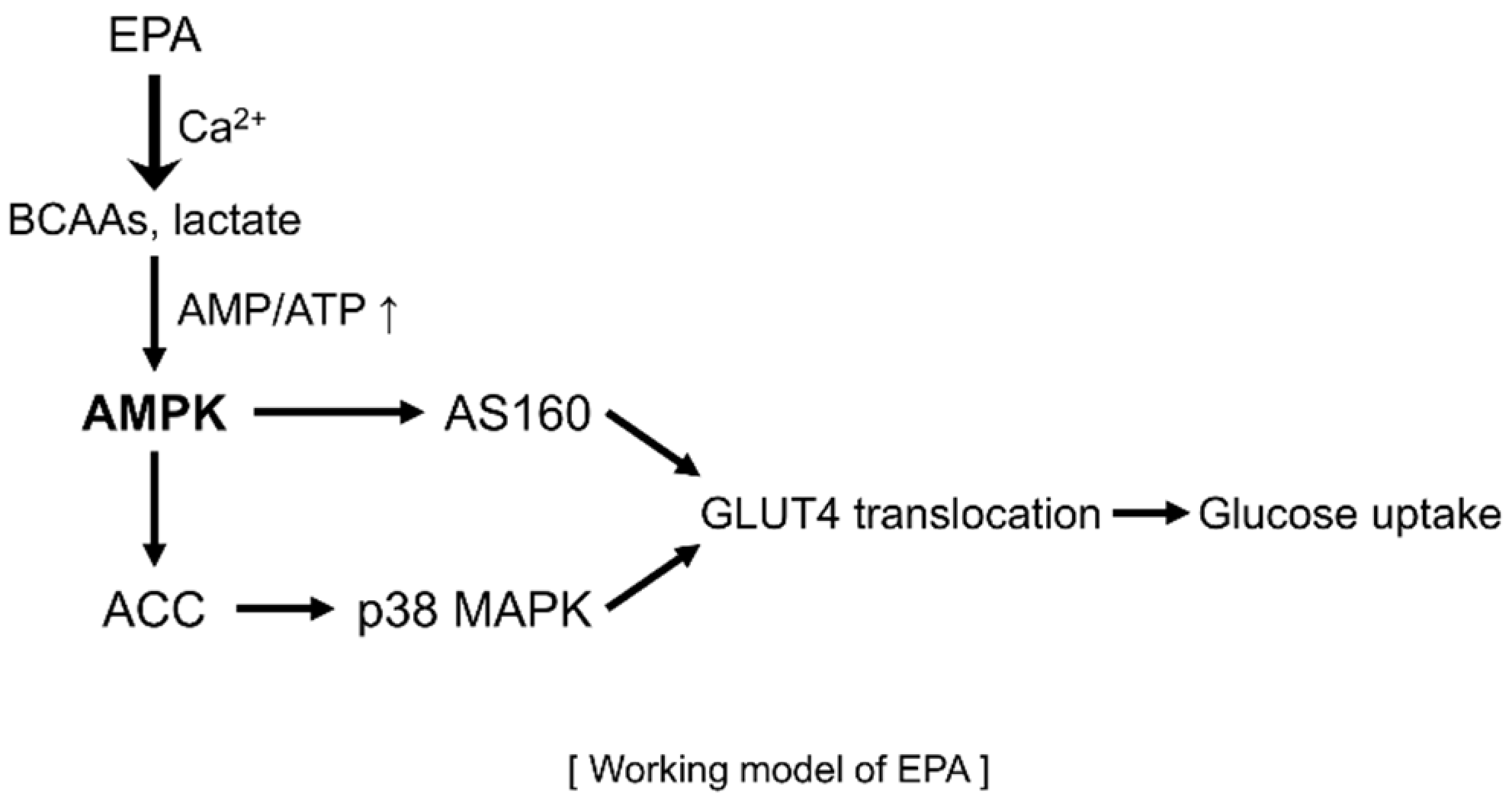
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Primary Myoblasts Preparation and Culture
4.4. NMR Analysis and Data Pre-Processing
4.5. Cellular Metabolic Rate
4.6. Western Blot Analysis
4.7. Assessment of Intracellular Calcium
4.8. Uptake of 2-deoxy-d(H3)-glucose
4.9. Myc-GLUT4 Translocation Assay
4.10. RT-Qpcr
4.11. Silencing of Genes Encoding AMPKα2 and p38 MAPK
4.12. Cytochemistry
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.T.; Kim, S.H.; Lee, M.S.; Kim, S.H.; Yang, H.J.; Kim, M.J.; Kim, H.S.; Ha, J.; Kim, M.S.; Kwon, D.Y. Anti-obesity effects of ginsenoside Rh2 are associated with the activation of AMPK signaling pathway in 3T3-L1 adipocyte. Biochem. Biophys. Res. Commun. 2007, 364, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Invest. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase as a drug target. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 185–210. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Huang, C.; Nawaz, Z.; Levy, M.; Somwar, R.; Li, D.; Bilan, P.J.; Klip, A. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J. Biol. Chem. 2003, 278, 17953–17962. [Google Scholar] [CrossRef]
- Russell, R.R., 3rd; Bergeron, R.; Shulman, G.I.; Young, L.H. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. 1999, 277, H643–H649. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 136, 207S–211S. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhang, C.; Dong, Y.; Wang, S.; Song, P.; Viollet, B.; Zou, M.H. Activation of the AMP-activated protein kinase by eicosapentaenoic acid (EPA, 20:5 n-3) improves endothelial function in vivo. PLoS ONE 2012, 7, e35508. [Google Scholar] [CrossRef] [PubMed]
- Perez-Matute, P.; Perez-Echarri, N.; Martinez, J.A.; Marti, A.; Moreno-Aliaga, M.J. Eicosapentaenoic acid actions on adiposity and insulin resistance in control and high-fat-fed rats: Role of apoptosis, adiponectin and tumour necrosis factor-alpha. Br. J. Nutr. 2007, 97, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cebrian, S.; Perez-Matute, P.; Martinez, J.A.; Marti, A.; Moreno-Aliaga, M.J. Effects of eicosapentaenoic acid (EPA) on adiponectin gene expression and secretion in primary cultured rat adipocytes. J. Physiol. Biochem. 2006, 62, 61–69. [Google Scholar] [PubMed]
- Perez-Echarri, N.; Perez-Matute, P.; Marcos-Gomez, B.; Martinez, J.A.; Moreno-Aliaga, M.J. Effects of eicosapentaenoic acid ethyl ester on visfatin and apelin in lean and overweight (cafeteria diet-fed) rats. Br. J. Nutr. 2009, 101, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Jacob, R.F. Eicosapentaenoic acid inhibits glucose-induced membrane cholesterol crystalline domain formation through a potent antioxidant mechanism. Biochim. Biophys. Acta 2015, 1848, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, J.Y.; Lee, W.H.; Tsai, Y.H.; Chen, C.Y.; Chao, S.Y.; Hsieh, R.H. Functional modulation of mitochondria by eicosapentaenoic acid provides protection against ceramide toxicity to C6 glioma cells. J. Agric. Food Chem. 2009, 57, 11455–11462. [Google Scholar] [CrossRef]
- Lee, M.-S.; Kim, I.-H.; Kim, Y. Effects of eicosapentaenoic acid and docosahexaenoic acid on uncoupling protein 3 gene expression in C(2)C(12) muscle cells. Nutrients 2013, 5, 1660–1671. [Google Scholar] [CrossRef]
- Huang, M.; Paglialunga, S.; Wong, J.M.; Hoang, M.; Pillai, R.; Joseph, J.W. Role of prolyl hydroxylase domain proteins in the regulation of insulin secretion. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef]
- Gonzalez-Barroso, M.M.; Anedda, A.; Gallardo-Vara, E.; Redondo-Horcajo, M.; Rodriguez-Sanchez, L.; Rial, E. Fatty acids revert the inhibition of respiration caused by the antidiabetic drug metformin to facilitate their mitochondrial beta-oxidation. Biochim. Biophys. Acta 2012, 1817, 1768–1775. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Chen, W.; London, R.E.; Murphy, E.; Steenbergen, C. Preconditioning enhanced glucose uptake is mediated by p38 MAP kinase not by phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 11981–11986. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Bryant, N.J.; Govers, R.; James, D.E. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Mîinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peränen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sarbolouki, S.; Javanbakht, M.H.; Derakhshanian, H.; Hosseinzadeh, P.; Zareei, M.; Hashemi, S.B.; Dorosty, A.R.; Eshraghian, M.R.; Djalali, M. Eicosapentaenoic acid improves insulin sensitivity and blood sugar in overweight type 2 diabetes mellitus patients: A double-blind randomised clinical trial. Singapore Med. J. 2013, 54, 387–390. [Google Scholar] [PubMed]
- Tonouchi, M.; Hatta, H.; Bonen, A. Muscle contraction increases lactate transport while reducing sarcolemmal MCT4, but not MCT1. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1062–E1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, S.O.; Hoogeveen, R.C.; Brancati, F.L.; Astor, B.C.; Ballantyne, C.M.; Schmidt, M.I.; Young, J.H. Association of blood lactate with type 2 diabetes: The Atherosclerosis Risk in Communities Carotid MRI Study. Int. J. Epidemiol. 2010, 39, 1647–1655. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bois, P.R.J.; Grosveld, G.C. FKHR (FOXO1a) is required for myotube fusion of primary mouse myoblasts. EMBO J. 2003, 22, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, T.; van den Berg, F.; Tomasi, G.; Bro, R. Automated alignment of chromatographic data. J. Chemom. 2006, 20, 484–497. [Google Scholar] [CrossRef]
- Wijesekara, N.; Tung, A.; Thong, F.; Klip, A. Muscle cell depolarization induces a gain in surface GLUT4 via reduced endocytosis independently of AMPK. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1276–E1286. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Kang, M.S.; Nam, M.; Kim, S.A.; Hwang, G.-S.; Kim, H.S. Eicosapentaenoic Acid (EPA) Modulates Glucose Metabolism by Targeting AMP-Activated Protein Kinase (AMPK) Pathway. Int. J. Mol. Sci. 2019, 20, 4751. https://doi.org/10.3390/ijms20194751
Kim N, Kang MS, Nam M, Kim SA, Hwang G-S, Kim HS. Eicosapentaenoic Acid (EPA) Modulates Glucose Metabolism by Targeting AMP-Activated Protein Kinase (AMPK) Pathway. International Journal of Molecular Sciences. 2019; 20(19):4751. https://doi.org/10.3390/ijms20194751
Chicago/Turabian StyleKim, Nami, Mi Sun Kang, Miso Nam, Shin Ae Kim, Geum-Sook Hwang, and Hyeon Soo Kim. 2019. "Eicosapentaenoic Acid (EPA) Modulates Glucose Metabolism by Targeting AMP-Activated Protein Kinase (AMPK) Pathway" International Journal of Molecular Sciences 20, no. 19: 4751. https://doi.org/10.3390/ijms20194751