YC-1 Prevents Tumor-Associated Tissue Factor Expression and Procoagulant Activity in Hypoxic Conditions by Inhibiting p38/NF-κB Signaling Pathway

Abstract
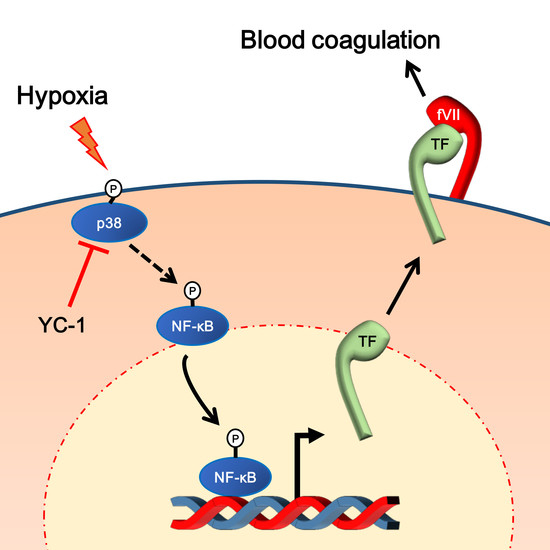
:
1. Introduction
2. Results
2.1. YC-1 Inhibits Hypoxia-Induced TF Expression in Human Cancer Cells
2.2. YC-1 Inhibits Hypoxia-Enhanced Procoagulant and Platelet-Stimulating Activity in A549 Cells
2.3. YC-1 Inhibits Hypoxia-Induced TF Via a HIF-1α-Independent Manner in A549 Cells
2.4. YC-1 Activates Cyclic Nucleotide-Dependent Protein Kinases in A549 Cells
2.5. YC-1 Inhibits Hypoxia-Induced NF-κB Activation
2.6. YC-1 Prevents Hypoxia-Induced TF through Inhibition of the p38/NF-κB Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Hypoxia Incubation
4.3. MTT Assay
4.4. Cell Surface TF Activity Assay
4.5. Measurement of Cancer-Induced Platelet Aggregation
4.6. Tilt Tube Plasma Clotting Assay
4.7. RNA Interference
4.8. Reverse Transcription Real-Time Polymerase Chain Reaction (PCR)
4.9. Western Blot Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Caine, G.J.; Stonelake, P.S.; Lip, G.Y.H.; Kehoe, S.T. The hypercoagulable state of malignancy: Pathogenesis and current debate. Neoplasia 2002, 4, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P.; Falanga, A.; Piccioli, A. Cancer and venous thromboembolism. Lancet Oncol. 2005, 6, 401–410. [Google Scholar] [CrossRef]
- Bach, R.R. Initiation of coagulation by tissue factor. CRC Crit. Rev. Biochem. 1988, 23, 339–368. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Connolly, G.C. Assessing risk of venous thromboembolism in the patient with cancer. J. Clin. Oncol. 2009, 27, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Callander, N.S.; Varki, N.; Rao, L.V. Immunohistochemical identification of tissue factor in solid tumors. Cancer. 1992, 70, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Escobar, I.; Beato-Zambrano, C.; Munoz Langa, J.; Brozos Vazquez, E.; Obispo Portero, B.; Gutierrez-Abad, D.; Munoz Martin, A.J. Pleiotropic effects of heparins: Does anticoagulant treatment increase survival in cancer patients? Clin. Transl. Oncol. 2018, 20, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Regina, S.; Valentin, J.B.; Lachot, S.; Lemarie, E.; Rollin, J.; Gruel, Y. Increased tissue factor expression is associated with reduced survival in non-small cell lung cancer and with mutations of TP53 and PTEN. Clin. Chem. 2009, 55, 1834–1842. [Google Scholar] [CrossRef]
- Mukai, M.; Oka, T. Mechanism and management of cancer-associated thrombosis. J. Cardiol. 2018, 72, 89–93. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Cui, X.Y.; Tinholt, M.; Stavik, B.; Dahm, A.E.; Kanse, S.; Jin, Y.; Seidl, S.; Sahlberg, K.K.; Iversen, N.; Skretting, G.; et al. Effect of hypoxia on tissue factor pathway inhibitor expression in breast cancer. J. Thromb. Haemost. 2016, 14, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Koizume, S.; Miyagi, Y. Tissue Factor-Factor VII Complex as a Key Regulator of Ovarian Cancer Phenotypes. Biomark. Cancer 2015, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Post, D.E.; Pieper, R.O.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005, 65, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, A.; Zakrzewicz, A.; Huber, K.; Thierbach, H.; Pepke, W.; Goldin-Lang, P.; Schultheiss, H.P.; Pries, A.; Rauch, U. Regulation of pro-angiogenic tissue factor expression in hypoxia-induced human lung cancer cells. Oncol. Rep. 2013, 30, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.; Asselin, M.C.; Reymen, B.; Jackson, A.; Lambin, P.; West, C.M.L.; O’Connor, J.P.B.; Faivre-Finn, C. Targeting hypoxia to improve mon-small cell lung cancer outcome. J. Natl. Cancer Inst. 2018, 110, 14–30. [Google Scholar] [CrossRef]
- Ko, F.N.; Wu, C.C.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1, a novel activator of platelet guanylate cyclase. Blood 1994, 84, 4226–4233. [Google Scholar]
- Wu, C.C.; Ko, F.N.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br. J. Pharmacol. 1995, 116, 1973–1978. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.M.; Wu, C.C.; Ko, F.N.; Lee, F.Y.; Kuo, S.C. YC-1, a nitric oxide-independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur. J. Pharmacol. 1997, 320, 161–166. [Google Scholar] [CrossRef]
- Chun, Y.S.; Yeo, E.J.; Park, J.W. Versatile pharmacological actions of YC-1: Anti-platelet to anticancer. Cancer Lett. 2004, 207, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Yeo, E.J.; Choi, E.; Teng, C.M.; Bae, J.M.; Kim, M.S.; Park, J.W. Inhibitory effect of YC-1 on the hypoxic induction of erythropoietin and vascular endothelial growth factor in Hep3B cells. Biochem. Pharmacol. 2001, 61, 947–954. [Google Scholar] [CrossRef]
- Yeo, E.J.; Chun, Y.S.; Cho, Y.S.; Kim, J.; Lee, J.C.; Kim, M.S.; Park, J.W. YC-1: A potential anticancer drug targeting hypoxia-inducible factor 1. J. Natl. Cancer Inst. 2003, 95, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.L.; Guh, J.H.; Peng, C.Y.; Wang, S.W.; Chang, Y.L.; Cheng, F.C.; Chang, J.H.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1 [3-(5’-hydroxymethyl-2’-furyl)-1-benzyl indazole] inhibits endothelial cell functions induced by angiogenic factors in vitro and angiogenesis in vivo models. J. Pharmacol. Exp. Ther. 2005, 314, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Ikezawa, Y.; Sakakibara-Konishi, J.; Mizugaki, H.; Oizumi, S.; Nishimura, M. Inhibition of Notch and HIF enhances the antitumor effect of radiation in Notch expressing lung cancer. Int. J. Clin. Oncol. 2017, 22, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Ryu, J.H.; Chun, Y.S.; Cho, Y.S.; Jang, I.J.; Cho, H.; Kim, J.; Kim, M.S.; Park, J.W. YC-1 induces S cell cycle arrest and apoptosis by activating checkpoint kinases. Cancer Res. 2006, 66, 6345–6352. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Kim, J.H.; Jung, Y.J.; Kim, K.E.; Jeong, J.M.; Chun, Y.S.; Park, J.W. Preclinical evaluation of YC-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett. 2007, 255, 107–116. [Google Scholar] [CrossRef]
- Chen, C.J.; Hsu, M.H.; Huang, L.J.; Yamori, T.; Chung, J.G.; Lee, F.Y.; Teng, C.M.; Kuo, S.C. Anticancer mechanisms of YC-1 in human lung cancer cell line, NCI-H226. Biochem. Pharmacol. 2008, 75, 360–368. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Wei, C.K.; Chang, F.R.; Hsieh, P.W.; Wu, C.C. Inhibition of the interactions between metastatic human breast cancer cells and platelets by beta-nitrostyrene derivatives. Life Sci. 2015, 143, 147–155. [Google Scholar] [CrossRef]
- Stasch, J.P.; Becker, E.M.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Feurer, A.; Gerzer, R.; Minuth, T.; Perzborn, E.; Pleiss, U.; et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 2001, 410, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Narita, I.; Shimada, M.; Yamabe, H.; Kinjo, T.; Tanno, T.; Nishizaki, K.; Kawai, M.; Nakamura, M.; Murakami, R.; Nakamura, N.; et al. NF-kappaB-dependent increase in tissue factor expression is responsible for hypoxic podocyte injury. Clin. Exp. Nephrol. 2016, 20, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Tuzovic, M.; Herrmann, J.; Iliescu, C.; Marmagkiolis, K.; Ziaeian, B.; Yang, E.H. Arterial thrombosis in patients with cancer. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The platelet lifeline to cancer: Challenges and opportunities. Cancer Cell. 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Amano, H.; Ito, Y.; Eshima, K.; Suzuki, T.; Ogawa, F.; Iyoda, A.; Satoh, Y.; Kato, S.; Nakamura, M.; et al. Thromboxane A(2) receptor signaling facilitates tumor colonization through P-selectin-mediated interaction of tumor cells with platelets and endothelial cells. Cancer Sci. 2012, 103, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Hu, F.; Huang, R.; Mackman, N.; Horowitz, J.M.; Jensen, R.L.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006, 66, 7067–7074. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Bendahl, P.O.; Belting, M.; Branco, C.; Johnson, R.S. Diverse roles of cell-specific hypoxia-inducible factor 1 in cancer-associated hypercoagulation. Blood 2016, 127, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Sovershaev, M.A.; Egorina, E.M.; Hansen, J.B.; Osterud, B.; Pacher, P.; Stasch, J.P.; Evgenov, O.V. Soluble guanylate cyclase agonists inhibit expression and procoagulant activity of tissue factor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1578–1586. [Google Scholar] [CrossRef]
- Hirata, Y.; Masuda, Y.; Kakutani, H.; Higuchi, T.; Takada, K.; Ito, A.; Nakagawa, Y.; Ishii, H. Sp1 is an essential transcription factor for LPS-induced tissue factor expression in THP-1 monocytic cells, and nobiletin represses the expression through inhibition of NF-kappaB, AP-1, and Sp1 activation. Biochem. Pharmacol. 2008, 75, 1504–1514. [Google Scholar] [CrossRef]
- Han, X.; Guo, B.; Li, Y.; Zhu, B. Tissue factor in tumor microenvironment: A systematic review. J. Hematol. Oncol. 2014, 7, 54. [Google Scholar] [CrossRef]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-κB. Mol. Cell. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.M.; Liang, Q.; Stafford, S.J.; Goplen, N.; Dharajiya, N.; Guo, L.; Sur, S.; Gaestel, M.; Alam, R. MK2 controls the level of negative feedback in the NF-kappaB pathway and is essential for vascular permeability and airway inflammation. J. Exp. Med. 2007, 204, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yang, C.X.; Zhang, B.; Wang, H.B.; Liu, H.Z.; Lai, X.H.; Liao, M.J.; Zhang, T. Sevoflurane suppresses hypoxia-induced growth and metastasis of lung cancer cells via inhibiting hypoxia-inducible factor-1alpha. J. Anesth. 2015, 29, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Tripathy, D.; Yin, X.; Desobry, K.; Martinez, J.; Riley, J.; Gay, D.; Luo, J.; Grammas, P. p38 MAPK: A mediator of hypoxia-induced cerebrovascular inflammation. J. Alzheimers Dis. 2012, 32, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Weerackody, R.P.; Welsh, D.J.; Wadsworth, R.M.; Peacock, A.J. Inhibition of p38 MAPK reverses hypoxia-induced pulmonary artery endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1312–H1320. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.; McNicholas, W.T.; Taylor, C.T. A critical role for p38 map kinase in NF-kappaB signaling during intermittent hypoxia/reoxygenation. Biochem. Biophys. Res. Commun. 2007, 355, 728–733. [Google Scholar] [CrossRef]
- Hazzalin, C.A.; Cano, E.; Cuenda, A.; Barratt, M.J.; Cohen, P.; Mahadevan, L.C. p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr. Biol. 1996, 6, 1028–1031. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| TF | 5′-GCCAGGAGAAAGGGGAAT-3′ | 5′-CAGTGCAATATAGCATTTGCAGTAGC-3′ |
| VEGF | 5′-TCGGGCCTCCGAAACCATGA-3′ | 5′-CCTGGTGAGAGATCTGGTTC-3′ |
| β-actin | 5′-TCACCCACACTGTGCCCATCTACGA-3′ | 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, K.-Y.; Wei, C.-K.; Wu, C.-C. YC-1 Prevents Tumor-Associated Tissue Factor Expression and Procoagulant Activity in Hypoxic Conditions by Inhibiting p38/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 244. https://doi.org/10.3390/ijms20020244
Hsieh K-Y, Wei C-K, Wu C-C. YC-1 Prevents Tumor-Associated Tissue Factor Expression and Procoagulant Activity in Hypoxic Conditions by Inhibiting p38/NF-κB Signaling Pathway. International Journal of Molecular Sciences. 2019; 20(2):244. https://doi.org/10.3390/ijms20020244
Chicago/Turabian StyleHsieh, Kan-Yen, Chien-Kei Wei, and Chin-Chung Wu. 2019. "YC-1 Prevents Tumor-Associated Tissue Factor Expression and Procoagulant Activity in Hypoxic Conditions by Inhibiting p38/NF-κB Signaling Pathway" International Journal of Molecular Sciences 20, no. 2: 244. https://doi.org/10.3390/ijms20020244