Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Triptolide Attenuates Isoproterenol-Induced Cardiac Fibrosis and Collagen Deposition in Mice
2.2. Triptolide Reduces Angiotensin-II-Induced Collagen Synthesis of CFs
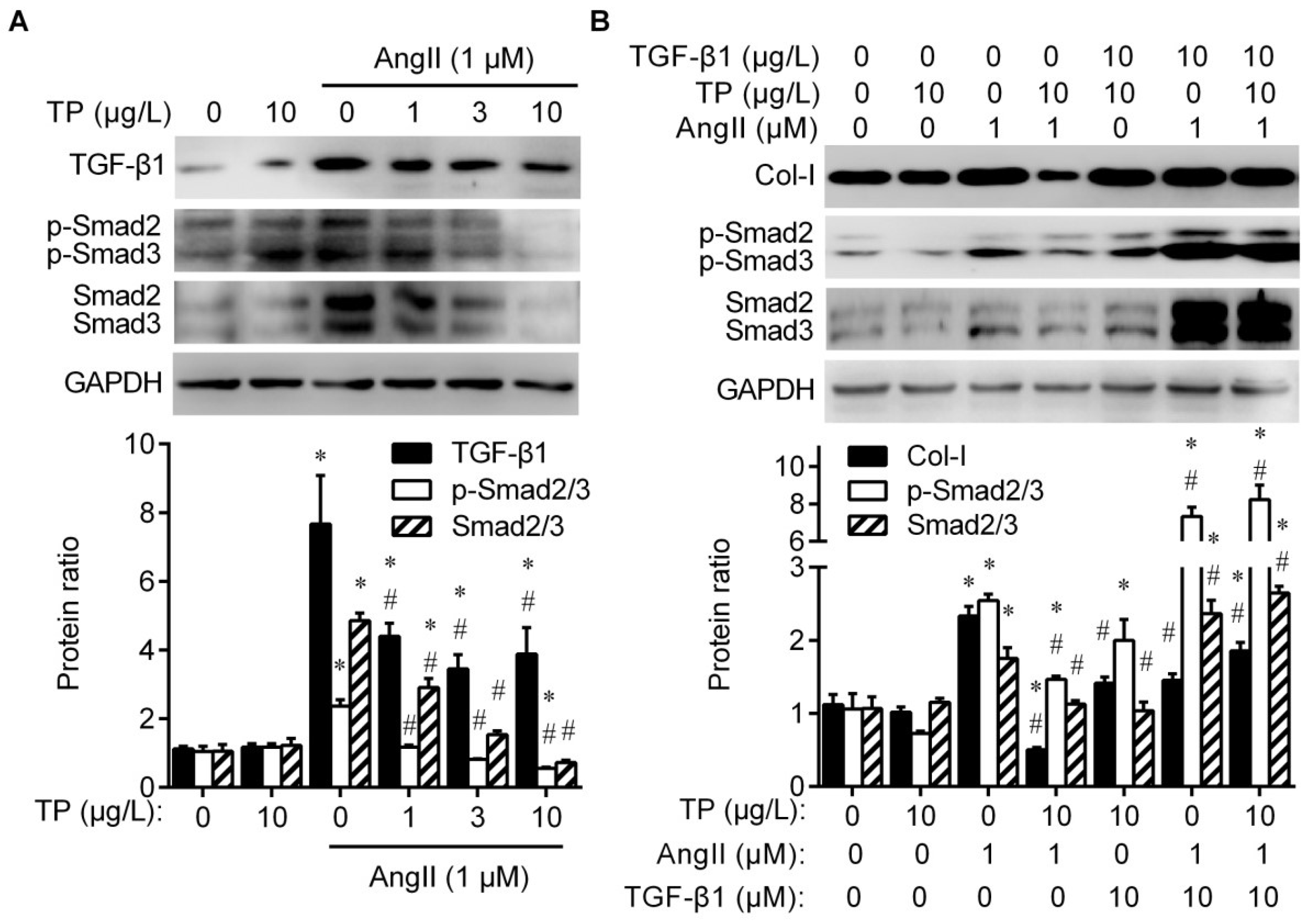
2.3. Triptolide Reduces Collagen Production by Inhibiting TGF-β1/Smad Signaling
2.4. Triptolide Inhibits Angiotensin II-Induced JNK and ERK1/2 Activation
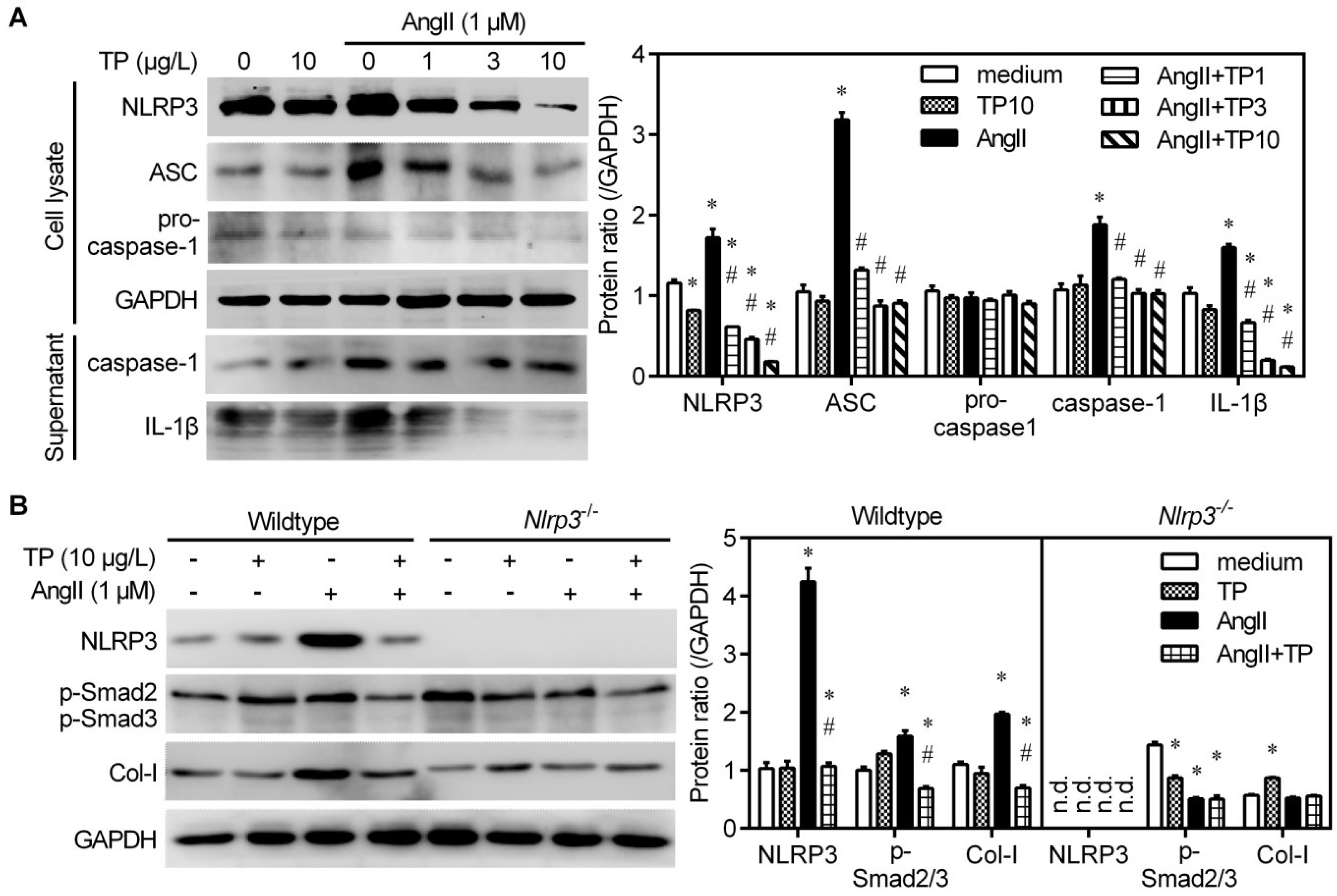
2.5. Triptolide Reduces Collagen Production by Inhibiting NLRP3 Inflammasome Activation
2.6. Triptolide Interrupts Angiotensin II-Induced Formation of the NLRP3 Inflammasome
2.7. Triptolide Inhibits Isoproterenol-Induced NLRP3-TGFβ1-Smad Signaling in the Mouse Ventricle
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Cell Culture
4.3. Histological Observations
4.4. Immunofluorescence
4.5. Western Blot
4.6. Colocalization
4.7. Immunoprecipitation
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, S.; Zhang, M.; Zhang, Z.; Hong, J.; Han, D.; Ma, J.; Zhang, S.B.; Okunieff, P.; Zhang, L. Triptolide mitigates radiation-induced pulmonary fibrosis via inhibition of axis of alveolar macrophages-NOXes-ROS-myofibroblasts. Cancer Biol. Ther. 2016, 17, 381–389. [Google Scholar] [CrossRef]
- Kania, G.; Blyszczuk, P.; Eriksson, U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc. Med. 2009, 19, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2017, 113, 5. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Mace, P.D.; Riedl, S.J. Structural mechanisms in NLR inflammasome signaling. Curr. Opin. Struct. Biol. 2014, 29, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M. The Role of the NLRP3 Inflammasome in Fibrosis. Open Rheumatol. J. 2012, 6, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.Y.; Li, J.M.; Guo, F.J.; Liu, Y.; Tong, Y.F.; Pan, X.C.; Lu, X.L.; Ye, W.; Chen, X.H.; Zhang, H.G. Triptolide Upregulates Myocardial Forkhead Helix Transcription Factor p3 Expression and Attenuates Cardiac Hypertrophy. Front. Pharmacol. 2016, 7, 471. [Google Scholar] [CrossRef]
- Liu, M.; Chen, J.; Huang, Y.; Ke, J.; Li, L.; Huang, D.; Wu, W. Triptolide alleviates isoprenaline-induced cardiac remodeling in rats via TGF-β1/Smad3 and p38 MAPK signaling pathway. Die Pharm. 2015, 70, 244–250. [Google Scholar]
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, X.; Ni, Y.; Zhang, K.; Dong, Z.; Yan, X.; Qin, J.; Sun, H.; Ding, Y.; Zhao, P.; et al. Triptolide protects rat heart against pressure overload-induced cardiac fibrosis. Int. J. Cardiol. 2013, 168, 2498–2505. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xue, M.; Li, C.J.; Yang, W.; Wang, S.S.; Ma, Z.J.; Zhang, X.N.; Wang, X.Y.; Zhao, R.; Chang, B.C.; et al. Protective effects of triptolide on TLR4 mediated autoimmune and inflammatory response induced myocardial fibrosis in diabetic cardiomyopathy. J. Ethnopharmacol. 2016, 193, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Wijekoon, C.P.; Liao, K.C.; Muruve, D.A. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life 2013, 65, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Ruland, J. Inflammasome: Putting the pieces together. Cell 2014, 156, 1127–1129. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Lindner, D.; Zietsch, C.; Tank, J.; Sossalla, S.; Fluschnik, N.; Hinrichs, S.; Maier, L.; Poller, W.; Blankenberg, S.; Schultheiss, H.P.; et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 2014, 109, 428. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 inflammasome as a novel player in myocardial infarction. Int. Heart J. 2014, 55, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Lee, M.E.; Jeong, J.; Lee, J.; Cho, S.; Seo, M.; Park, S. sRAGE attenuates angiotensin II-induced cardiomyocyte hypertrophy by inhibiting RAGE-NFκB-NLRP3 activation. Inflamm. Res. 2018, 67, 691–701. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, T.; Usui, F.; Kobayashi, M.; Komada, T.; Kimura, H.; Kawashima, A.; Ohkuchi, A.; Taniguchi, S.; Takahashi, M. NLRP3 Deficiency Improves Angiotensin II-Induced Hypertension But Not Fetal Growth Restriction During Pregnancy. Endocrinology 2015, 156, 4281–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Ziaei, S.; Halaby, R. Immunosuppressive, anti-inflammatory and anti-cancer properties of triptolide: A mini review. Avicenna J. Phytomed. 2016, 6, 149–164. [Google Scholar] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.F.; Wang, Y.; Ding, Y.Y.; Li, J.M.; Pan, X.C.; Lu, X.L.; Chen, X.H.; Liu, Y.; Zhang, H.G. Cyclin-Dependent Kinase Inhibitor p21WAF1/CIP1 Facilitates the Development of Cardiac Hypertrophy. Cell. Physiol. Biochem. 2017, 42, 1645–1656. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.-C.; Liu, Y.; Cen, Y.-Y.; Xiong, Y.-L.; Li, J.-M.; Ding, Y.-Y.; Tong, Y.-F.; Liu, T.; Chen, X.-H.; Zhang, H.-G. Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis. Int. J. Mol. Sci. 2019, 20, 360. https://doi.org/10.3390/ijms20020360
Pan X-C, Liu Y, Cen Y-Y, Xiong Y-L, Li J-M, Ding Y-Y, Tong Y-F, Liu T, Chen X-H, Zhang H-G. Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis. International Journal of Molecular Sciences. 2019; 20(2):360. https://doi.org/10.3390/ijms20020360
Chicago/Turabian StylePan, Xi-Chun, Ya Liu, Yan-Yan Cen, Ya-Lan Xiong, Jing-Mei Li, Yuan-Yuan Ding, Yang-Fei Tong, Tao Liu, Xiao-Hong Chen, and Hai-Gang Zhang. 2019. "Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis" International Journal of Molecular Sciences 20, no. 2: 360. https://doi.org/10.3390/ijms20020360
APA StylePan, X. -C., Liu, Y., Cen, Y. -Y., Xiong, Y. -L., Li, J. -M., Ding, Y. -Y., Tong, Y. -F., Liu, T., Chen, X. -H., & Zhang, H. -G. (2019). Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis. International Journal of Molecular Sciences, 20(2), 360. https://doi.org/10.3390/ijms20020360