Trans-Kingdom Conjugation within Solid Media from Escherichia coli to Saccharomyces cerevisiae

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
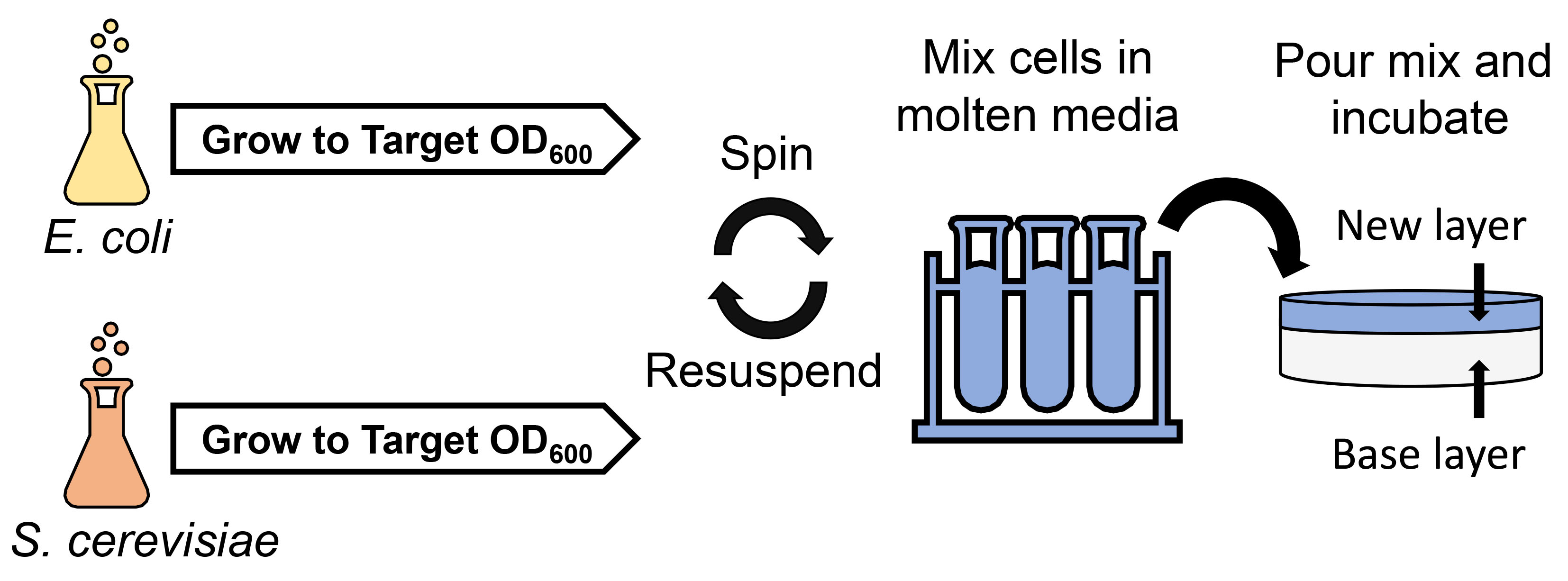
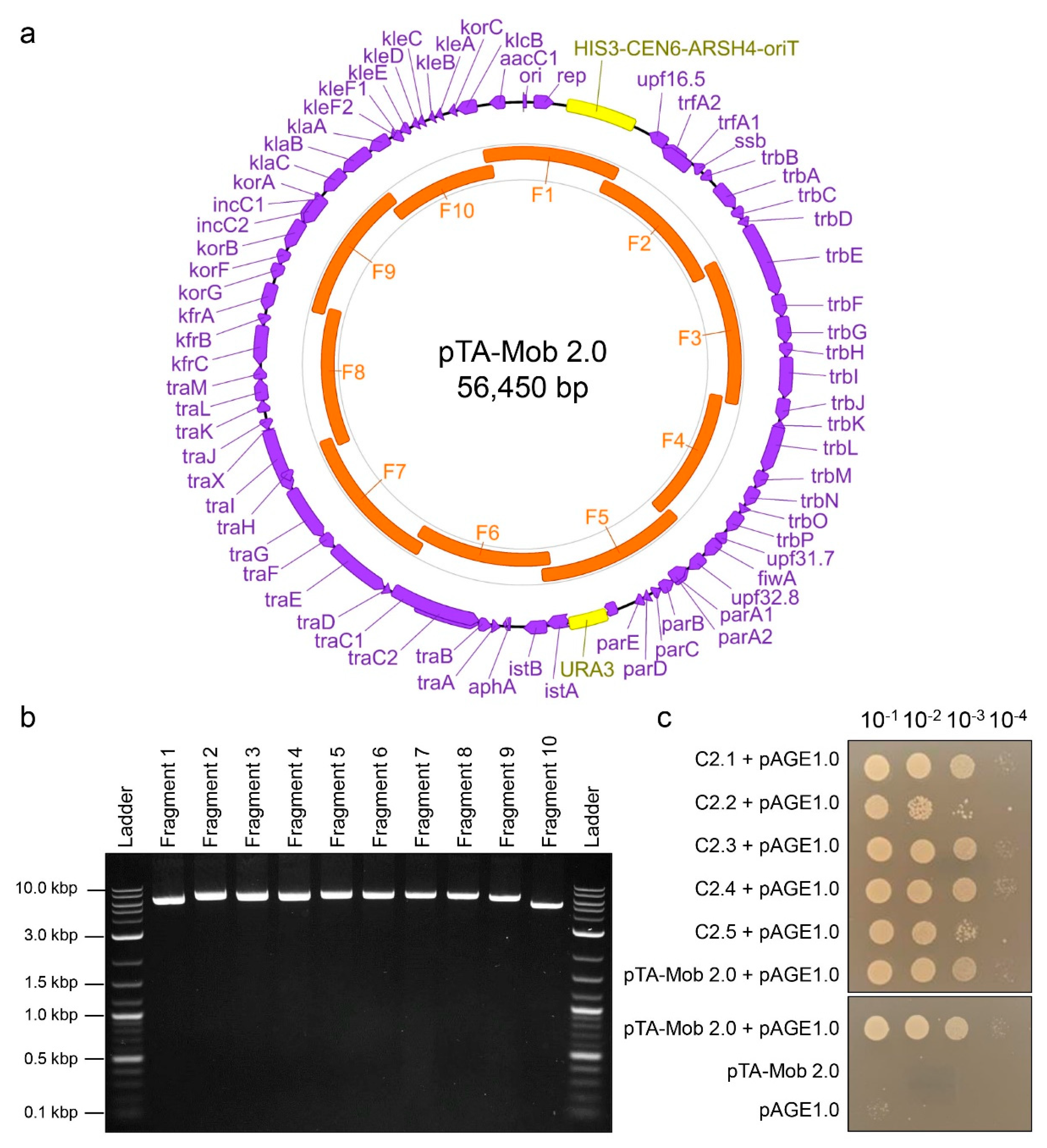
2.1. PCR-Based Synthesis of Conjugative Plasmid pTA-Mob 2.0
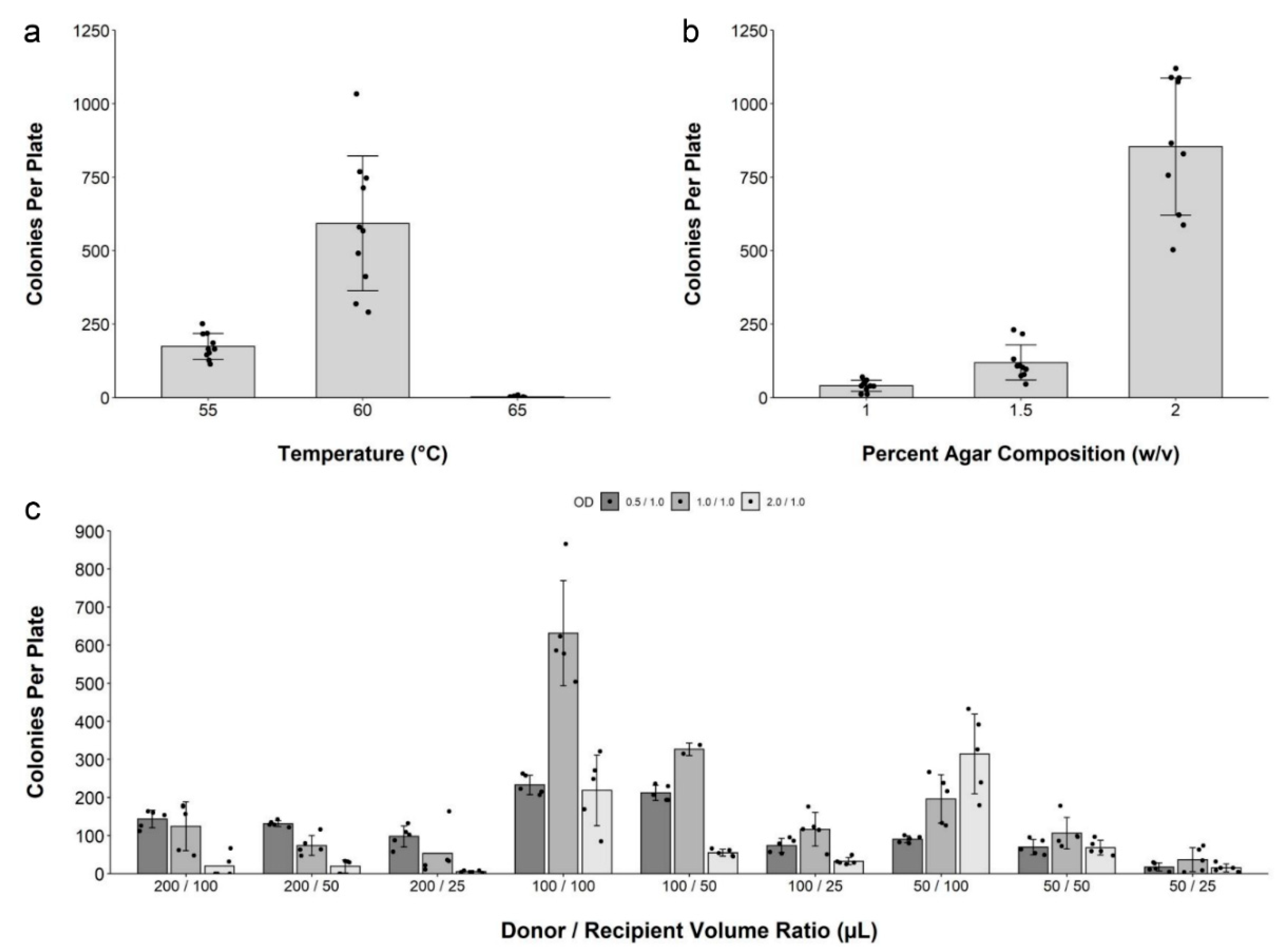
2.2. Development and Optimization of Conjugation within Solid Media
2.3. Conjugation of IncP-Based Plasmids of Increasing Size
3. Discussion
4. Materials and Methods
4.1. Strains and Growth Conditions
4.2. Construction of pTA-Mob 2.0 Plasmid
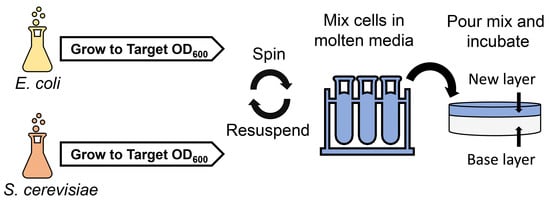
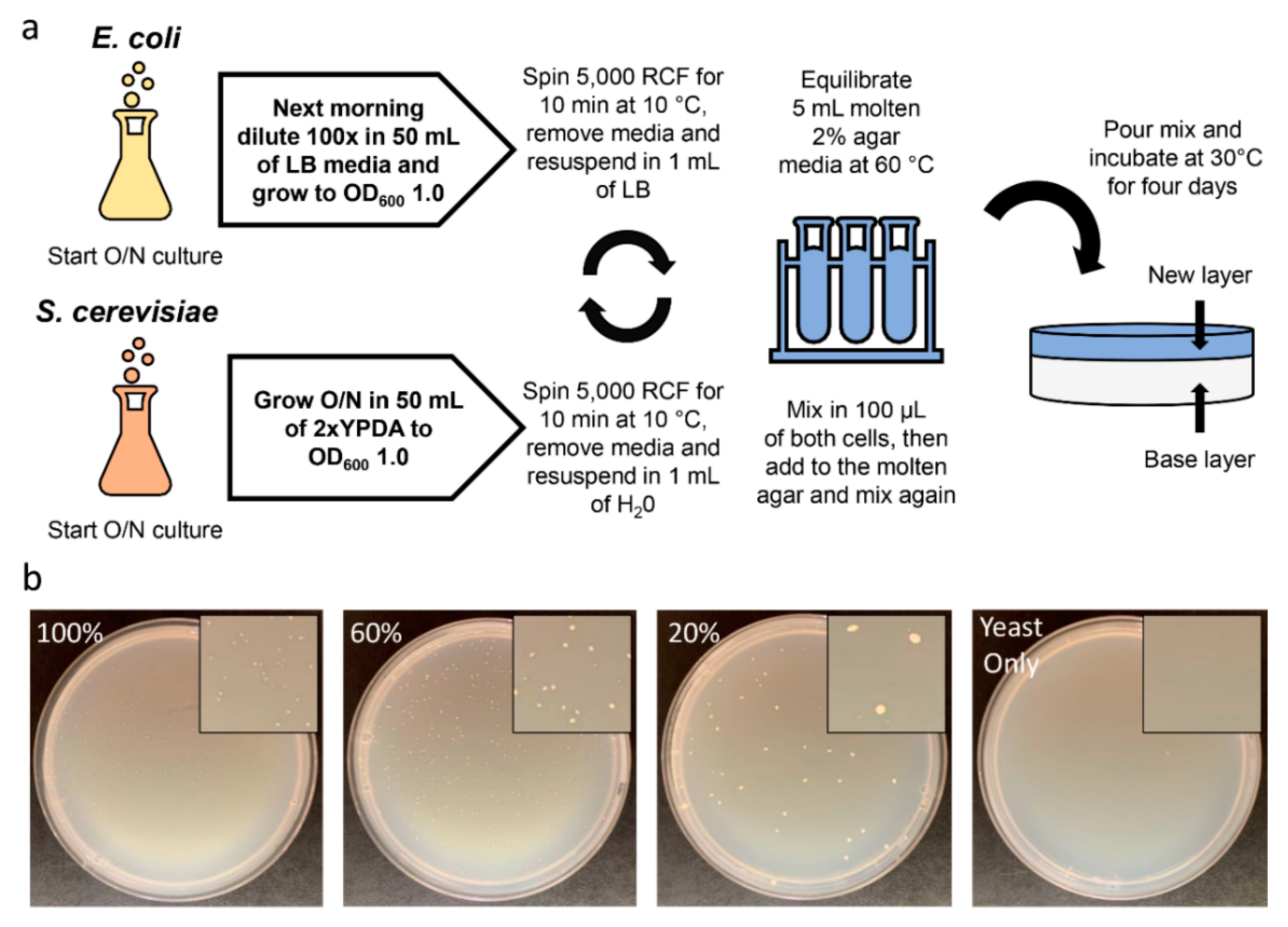
4.3. Conjugation within Solid Media
4.3.1. Preparation of E. coli
4.3.2. Preparation of S. cerevisiae
4.3.3. Conjugation within Solid Media Procedure
4.3.4. Confirmation of Successfully Transferred Plasmids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cabezón, E.; Ripoll-Rozada, J.; Peña, A.; De La Cruz, F.; Arechaga, I. Towards an integrated model of bacterial conjugation. FEMS Microbiol. Rev. 2014, 39, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilton, M.-D.; Drummond, M.H.; Merlo, D.J.; Sciaky, D.; Montoya, A.L.; Gordon, M.P.; Nester, E.W. Stable incorporation of plasmid DNA into higher plant cells: The molecular basis of crown gall tumorigenesis. Cell 1977, 11, 263–271. [Google Scholar] [CrossRef]
- Christie, P.J. Agrobacterium tumefaciens T-complex transport apparatus: A paradigm for a new family of multifunctional transporters in eubacteria. J. Bacteriol. 1997, 179, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.E.; Zambryski, P.C. Generic trans-kingdom sex? Nature 1989, 340, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, J.A.; Sprague, G.F. Bacterial conjugative plasmids mobilize DNA transfer between bacteria and yeast. Nature 1989, 340, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bundock, P.; den Dulk-Ras, A.; Beijersbergen, A.; Hooykaas, P.J. Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J. 1995, 14, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Cashmore, A.M.; Wilkins, B.M. IncP plasmids are unusually effective in mediating conjugation of Escherichia coli and Saccharomyces cerevisiae: Involvement of the tra2 mating system. J. Bacteriol. 1998, 180, 6538–6543. [Google Scholar]
- Moriguchi, K.; Yamamoto, S.; Ohmine, Y.; Suzuki, K. A Fast and Practical Yeast Transformation Method Mediated by Escherichia coli Based on a Trans-Kingdom Conjugal Transfer System: Just Mix Two Cultures and Wait One Hour. PLoS ONE 2016, 11, e0148989. [Google Scholar] [CrossRef]
- Brumwell, S.L.; MacLeod, M.R.; Huang, T.; Cochrane, R.R.; Meaney, R.S.; Zamani, M.; Matysiakiewicz, O.; Dan, K.N.; Janakirama, P.; Edgell, D.R.; et al. Designer Sinorhizobium meliloti strains and multi-functional vectors enable direct inter-kingdom DNA transfer. PLoS ONE 2019, 14, e0206781. [Google Scholar] [CrossRef]
- Karas, B.J.; Diner, R.E.; Lefebvre, S.C.; McQuaid, J.; Phillips, A.P.R.; Noddings, C.M.; Brunson, J.K.; Valas, R.E.; Deerinck, T.J.; Jablanovic, J.; et al. Designer diatom episomes delivered by bacterial conjugation. Nat. Commun. 2015, 6, 6925. [Google Scholar] [CrossRef]
- Diner, R.E.; Bielinski, V.A.; Dupont, C.L.; Allen, A.E.; Weyman, P.D. Refinement of the Diatom Episome Maintenance Sequence and Improvement of Conjugation-Based DNA Delivery Methods. Front. Bioeng. Biotechnol. 2016, 4, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, S.S.; Diamond, A.; Wang, H.; Therrien, J.A.; Lant, J.T.; Jazey, T.; Lee, K.; Klassen, Z.; Desgagné-Penix, I.; Karas, B.J.; et al. An Expanded Plasmid-Based Genetic Toolbox Enables Cas9 Genome Editing and Stable Maintenance of Synthetic Pathways in Phaeodactylum tricornutum. ACS Synth. Biol. 2018, 7, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Slattery, S.; Karas, B.; Edgell, D. Delivery of the Cas9 or TevCas9 System into Phaeodactylum tricornutum via Conjugation of Plasmids from a Bacterial Donor. Bio-Protocol 2018, 8, e2974. [Google Scholar] [CrossRef]
- Waters, V.L. Conjugation between bacterial and mammalian cells. Nat. Genet. 2001, 29, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Kunik, T.; Tzfira, T.; Kapulnik, Y.; Gafni, Y.; Dingwall, C.; Citovsky, V. Genetic transformation of HeLa cells by Agrobacterium. Proc. Natl. Acad. Sci. USA 2001, 98, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Fernández-González, E.; de Paz, H.D.; Alperi, A.; Agúndez, L.; Faustmann, M.; Sangari, F.J.; Dehio, C.; Llosa, M. Transfer of R388 derivatives by a pathogenesis-associated type IV secretion system into both bacteria and human cells. J. Bacteriol. 2011, 193, 6257–6265. [Google Scholar] [CrossRef] [PubMed]
- Schröder, G.; Schuelein, R.; Quebatte, M.; Dehio, C. Conjugative DNA transfer into human cells by the VirB/VirD4 type IV secretion system of the bacterial pathogen Bartonella henselae. Proc. Natl. Acad. Sci. USA 2011, 108, 14643–14648. [Google Scholar] [CrossRef]
- Aune, T.E.V.; Aachmann, F.L. Methodologies to increase the transformation efficiencies and the range of bacteria that can be transformed. Appl. Microbiol. Biotechnol. 2010, 85, 1301–1313. [Google Scholar] [CrossRef]
- Heinze, S.; Kornberger, P.; Grätz, C.; Schwarz, W.H.; Zverlov, V.V.; Liebl, W. Transmating: Conjugative transfer of a new broad host range expression vector to various Bacillus species using a single protocol. BMC Microbiol. 2018, 18, 56. [Google Scholar] [CrossRef]
- Karas, B.J.; Jablanovic, J.; Sun, L.; Ma, L.; Goldgof, G.M.; Stam, J.; Ramon, A.; Manary, M.J.; Winzeler, E.A.; Venter, J.C.; et al. Direct transfer of whole genomes from bacteria to yeast. Nat. Methods 2013, 10, 410–412. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.E.; Top, E.M. Gene transfer in soil systems using microcosms. FEMS Microbiol. Ecol. 1998, 25, 319–329. [Google Scholar] [CrossRef]
- Brophy, J.A.N.; Triassi, A.J.; Adams, B.L.; Renberg, R.L.; Stratis-Cullum, D.N.; Grossman, A.D.; Voigt, C.A. Engineered integrative and conjugative elements for efficient and inducible DNA transfer to undomesticated bacteria. Nat. Microbiol. 2018, 3, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Ronda, C.; Chen, S.P.; Cabral, V.; Yaung, S.J.; Wang, H.H. Metagenomic engineering of the mammalian gut microbiome in situ. Nat. Methods 2019, 16, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.A.; Pellegrino, G.M.; Therrien, J.A.; Ham, D.T.; Bartlett, P.C.; Karas, B.J.; Gloor, G.B.; Edgell, D.R. Efficient inter-species conjugative transfer of a CRISPR nuclease for targeted bacterial killing. Nat. Commun. 2019, 10, 4544. [Google Scholar] [CrossRef]
- Itaya, M.; Sato, M.; Hasegawa, M.; Kono, N.; Tomita, M.; Kaneko, S. Far rapid synthesis of giant DNA in the Bacillus subtilis genome by a conjugation transfer system. Sci. Rep. 2018, 8, 8792. [Google Scholar] [CrossRef]
- Zechner, E.L.; Moncalián, G.; de la Cruz, F. Relaxases and Plasmid Transfer in Gram-Negative Bacteria. In Type IV Secretion in Gram-Negative and Gram-Positive Bacteria; Backert, S., Grohmann, E., Eds.; Springer: Berlin, Germany, 2018; Volume 413, pp. 93–113. ISBN 978-3-319-75241-9. [Google Scholar]
- Christie, P.J. Type IV secretion: Intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol. Microbiol. 2001, 40, 294–305. [Google Scholar] [CrossRef]
- Bradley, D.E.; Taylor, D.E.; Cohen, D.R. Specification of surface mating systems among conjugative drug resistance plasmids in Escherichia coli K-12. J. Bacteriol. 1980, 143, 1466–1470. [Google Scholar] [Green Version]
- Mason, J.H. Isolation of anaerobic bacteria by a modified shake method. J. Gen. Microbiol. 1953, 8, 263–264. [Google Scholar] [CrossRef]
- Nagasaki, K.; Imai, I. Solid-phase culture of marine phytoflagellates. Bull. Jpn. Soc. Mircobial Ecol. 1994, 9, 37–43. [Google Scholar] [CrossRef]
- Lakeman, M.B.; Cattolico, A.R.A. Cryptic Diversity in Phytoplankton Cultures is Revealed Using a Simple Plating Technique. J. Phycol. 2007, 43, 662–674. [Google Scholar] [CrossRef]
- Koch, A.L. Growth Measurement. In Methods for General and Molecular Biology; Gerhard, P., Murray, R.G.E., Wood, W.A., Krieg, N.R., Eds.; American Society for Microbiology Press: Washington, DC, USA, 2007; pp. 172–199. [Google Scholar]
- Zhu, Y.O.; Sherlock, G.; Petrov, D.A. Whole Genome Analysis of 132 Clinical Saccharomyces cerevisiae Strains Reveals Extensive Ploidy Variation. G3 Genes Genomes Genet. 2016, 6, 2421–2434. [Google Scholar]
- Gajdács, M.; Ábrók, M.; Lázár, A.; Burián, K. Comparative Epidemiology and Resistance Trends of Common Urinary Pathogens in a Tertiary-Care Hospital: A 10-Year Surveillance Study. Medicina 2019, 55, 356. [Google Scholar] [Green Version]
- Strand, T.A.; Lale, R.; Degnes, K.F.; Lando, M.; Valla, S. A new and improved host-independent plasmid system for RK2-based conjugal transfer. PLoS ONE 2014, 9, e90372. [Google Scholar] [CrossRef] [PubMed]
- Pansegrau, W.; Lanka, E.; Barth, P.T.; Figurski, D.H.; Guiney, D.G.; Haas, D.; Helinski, D.R.; Schwab, H.; Stanisich, V.A.; Thomas, C.M. Complete Nucleotide Sequence of Birmingham IncPα Plasmids: Compilation and Comparative Analysis. J. Mol. Biol. 1994, 239, 623–663. [Google Scholar] [CrossRef]
- Karas, B.J.; Moreau, N.G.; Deerinck, T.J.; Gibson, D.G.; Venter, J.C.; Smith, H.O.; Glass, J.I. Direct Transfer of a Mycoplasma mycoides Genome to Yeast Is Enhanced by Removal of the Mycoides Glycerol Uptake Factor Gene glpF. ACS Synth. Biol. 2019, 8, 239–244. [Google Scholar] [CrossRef]
- Karas, B.J.; Molparia, B.; Jablanovic, J.; Hermann, W.J.; Lin, Y.-C.; Dupont, C.L.; Tagwerker, C.; Yonemoto, I.T.; Noskov, V.N.; Chuang, R.-Y.; et al. Assembly of eukaryotic algal chromosomes in yeast. J. Biol. Eng. 2013, 7, 30. [Google Scholar] [CrossRef]
- Miyano, M.; Tanaka, K.; Ishikawa, S.; Takenaka, S.; Miguel-Arribas, A.; Meijer, W.J.J.; Yoshida, K.I. Rapid conjugative mobilization of a 100kb segment of Bacillus subtilis chromosomal DNA is mediated by a helper plasmid with no ability for self-transfer. Microb. Cell Fact. 2018, 17, 13. [Google Scholar] [CrossRef]
- Moriguchi, K.; Yamamoto, S.; Tanaka, K.; Kurata, N.; Suzuki, K. Trans-Kingdom Horizontal DNA Transfer from Bacteria to Yeast Is Highly Plastic Due to Natural Polymorphisms in Auxiliary Nonessential Recipient Genes. PLoS ONE 2013, 8, e74590. [Google Scholar] [CrossRef]
- Quandt, J.; Clark, R.G.; Venter, A.P.; Clark, S.R.; Twelker, S.; Hynes, M.F. Modified RP4 and Tn5-Mob derivatives for facilitated manipulation of large plasmids in Gram-negative bacteria. Plasmid 2004, 52, 1–12. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, T.; Jia, J.; Guo, L.; Zhang, H.; Li, C.; Qiao, R. Establishment of a highly efficient conjugation protocol for Streptomyces kanamyceticus ATCC12853. MicrobiologyOpen 2019, 8, e00747. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Ardeshna, D.; Lin, J. Heat Shock-Enhanced Conjugation Efficiency in Standard Campylobacter jejuni Strains. Appl. Environ. Microbiol. 2015, 81, 4546–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, J.A.; Fagan, R.P. Heat shock increases conjugation efficiency in Clostridium difficile. Anaerobe 2016, 42, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, J.A.; Sprague, G.F. Transmission of Plasmid DNA to Yeast by Conjugation with Bacteria. Methods Enzymol. 1991, 194, 187–195. [Google Scholar] [PubMed]
- Karas, B.J.; Jablanovic, J.; Irvine, E.; Sun, L.; Ma, L.; Weyman, P.D.; Gibson, D.G.; Glass, J.I.; Venter, J.C.; Hutchison, C.A.; et al. Transferring whole genomes from bacteria to yeast spheroplasts using entire bacterial cells to reduce DNA shearing. Nat. Protoc. 2014, 9, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Diner, R.E.; Noddings, C.M.; Lian, N.C.; Kang, A.K.; McQuaid, J.B.; Jablanovic, J.; Espinoza, J.L.; Nguyen, N.A.; Anzelmatti, M.A.; Jansson, J.; et al. Diatom centromeres suggest a mechanism for nuclear DNA acquisition. Proc. Natl. Acad. Sci. USA 2017, 114, E6015–E6024. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltysiak, M.P.M.; Meaney, R.S.; Hamadache, S.; Janakirama, P.; Edgell, D.R.; Karas, B.J. Trans-Kingdom Conjugation within Solid Media from Escherichia coli to Saccharomyces cerevisiae. Int. J. Mol. Sci. 2019, 20, 5212. https://doi.org/10.3390/ijms20205212
Soltysiak MPM, Meaney RS, Hamadache S, Janakirama P, Edgell DR, Karas BJ. Trans-Kingdom Conjugation within Solid Media from Escherichia coli to Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2019; 20(20):5212. https://doi.org/10.3390/ijms20205212
Chicago/Turabian StyleSoltysiak, Maximillian P. M., Rebecca S. Meaney, Samir Hamadache, Preetam Janakirama, David R. Edgell, and Bogumil J. Karas. 2019. "Trans-Kingdom Conjugation within Solid Media from Escherichia coli to Saccharomyces cerevisiae" International Journal of Molecular Sciences 20, no. 20: 5212. https://doi.org/10.3390/ijms20205212