Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap)


{kind=link}
{kind=link}
Abstract
:1. Introduction
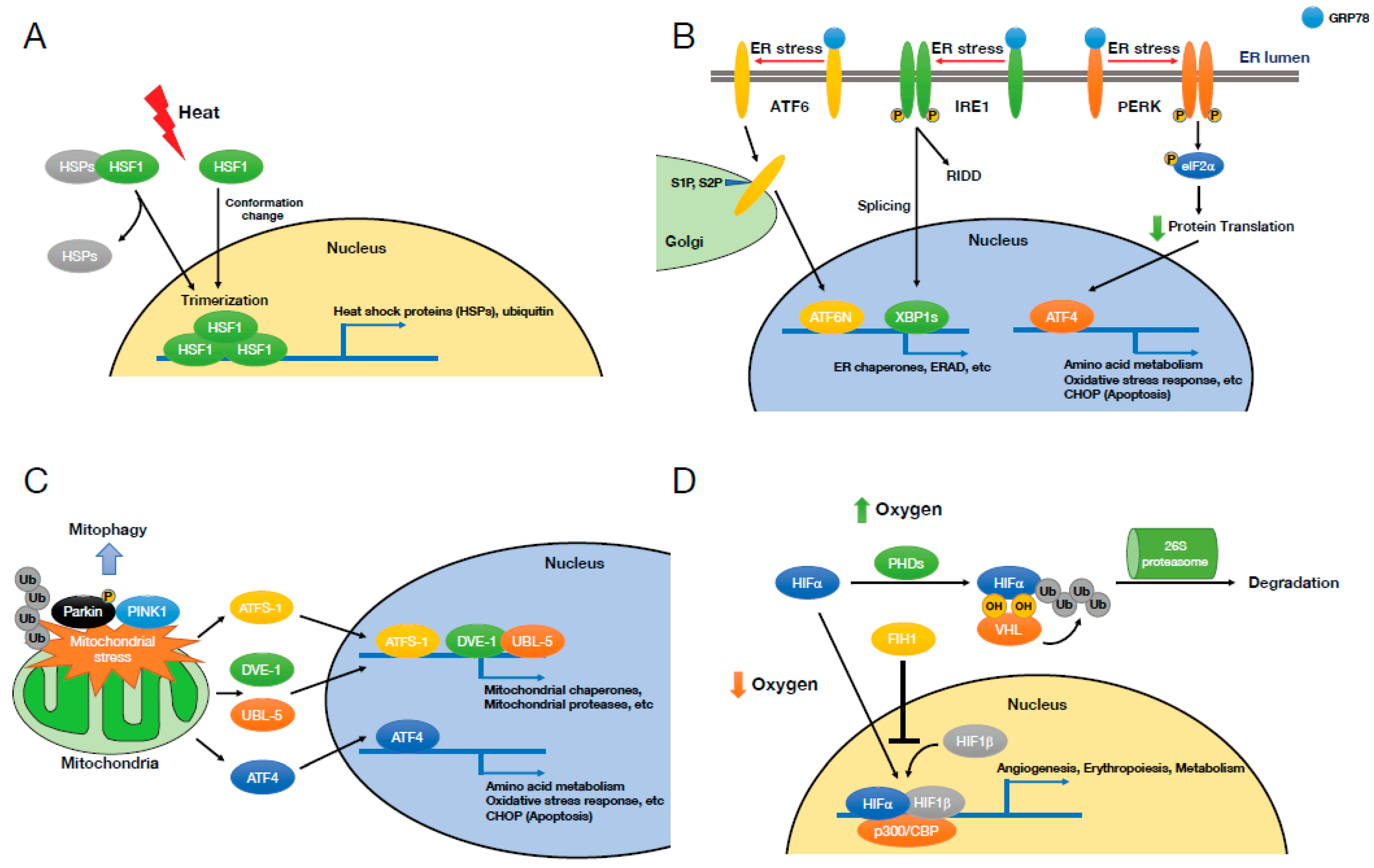
2. Cellular Stresses
2.1. Heat Shock Stress and Heat Shock Response
2.2. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR)
2.3. Mitochondrial Stress
2.4. Hypoxia
2.5. Oxidative Stress
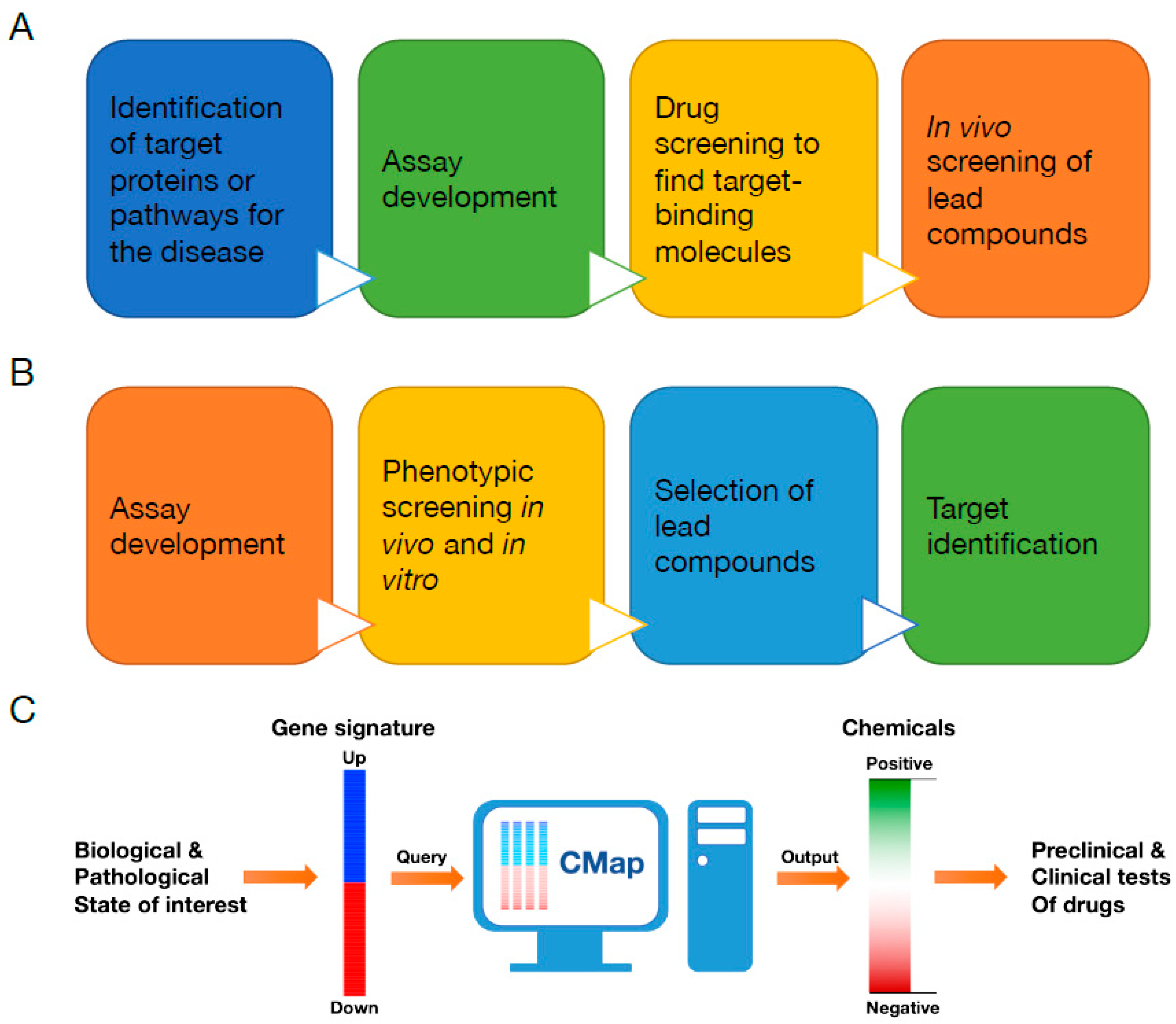
3. Connectivity Map (CMap)
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| β-TrCP | Beta-transducin repeat containing E3 ubiquitin protein ligase |
| AAA | ATPases associated with diverse cellular activities |
| AFG3L2 | AFG3-like protein 2 |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| BNIP3L | BCL2 interacting protein 3 like |
| BRD7 | Bromodomain-containing protein 7 |
| CLPP | Clp protease proteolytic subunit |
| CUL1 | Cullin 1 |
| CUL3 | Cullin 3 |
| ERK | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration, USA |
| HRD1 | HMG-CoA reductase degradation 1 homolog |
| IDH2 | Isocitrate dehydrogenase 2 |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase subunit beta |
| JNK | c-Jun N-terminal kinase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| MAPK | Mitogen-activated protein kinase |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced form |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| PI3K | Phosphoinositide 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| RBX1 | Ring box 1 |
| SKP1 | S-phase kinase-associated protein 1 |
| sMAF | Small musculoaponeurotic fibrosarcoma |
| SPG7 | Spastic paraplegia 7 |
| TCA | Tricarboxylic acid |
| VEGF-B | Vascular endothelial growth factor B |
References
- Vijg, J.; Le Bourg, E. Aging and the Inevitable Limit to Human Life Span. Gerontology 2017, 63, 432–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef]
- Lee, J.; Ozcan, U. Unfolded protein response signaling and metabolic diseases. J. Biol. Chem. 2014, 289, 1203–1211. [Google Scholar] [CrossRef]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsky, J.L.; Chiosis, G. Adapting to stress—Chaperome networks in cancer. Nat. Rev. Cancer 2018, 18, 562–575. [Google Scholar] [CrossRef]
- Buono, R.; Longo, V.D. Starvation, Stress Resistance, and Cancer. Trends Endocrinol. Metab. 2018, 29, 271–280. [Google Scholar] [CrossRef]
- Steward, W.P.; Brown, K. Cancer chemoprevention: A rapidly evolving field. Br. J. Cancer 2013, 109, 1–7. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Moran Luengo, T.; Mayer, M.P.; Rudiger, S.G.D. The Hsp70-Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 2019, 29, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Kregel, K.C. Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance. J. Appl. Physiol. 1985 2002, 92, 2177–2186. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Javid, B.; MacAry, P.A.; Lehner, P.J. Structure and function: Heat shock proteins and adaptive immunity. J. Immunol. 2007, 179, 2035–2040. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Mercier, P.A.; Winegarden, N.A.; Westwood, J.T. Human heat shock factor 1 is predominantly a nuclear protein before and after heat stress. J. Cell Sci. 1999, 112, 2765–2774. [Google Scholar] [PubMed]
- Baler, R.; Dahl, G.; Voellmy, R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol. Cell. Biol. 1993, 13, 2486–2496. [Google Scholar] [CrossRef]
- Sarge, K.D.; Murphy, S.P.; Morimoto, R.I. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol. Cell. Biol. 1993, 13, 1392–1407. [Google Scholar] [CrossRef]
- Ali, A.; Bharadwaj, S.; O’Carroll, R.; Ovsenek, N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol. Cell. Biol. 1998, 18, 4949–4960. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef]
- Hentze, N.; Le Breton, L.; Wiesner, J.; Kempf, G.; Mayer, M.P. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. Elife 2016, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.F.; Morimoto, R.I. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 2004, 15, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Takaki, E.; Hayashi, T.; Kitaura, Y.; Tanaka, Y.; Inouye, S.; Nakai, A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 2005, 280, 34908–34916. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, N.; Fujimoto, M.; Tan, K.; Prakasam, R.; Shinkawa, T.; Li, L.; Ichikawa, H.; Takii, R.; Nakai, A. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010, 29, 3459–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The heat shock response plays an important role in TDP-43 clearance: Evidence for dysfunction in amyotrophic lateral sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.; Trojanowski, J.Q.; Lee, V.M.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [PubMed]
- Ali, M.M.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.P.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Chapman, R.E.; Walter, P. Translational attenuation mediated by an mRNA intron. Curr. Biol. 1997, 7, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, T.; Yanagi, H.; Yura, T.; Mori, K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 1997, 8, 1845–1862. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Salazar Hernandez, M.A.; Auen, T.; Mucka, P.; Lee, J.; Ozcan, U. PGC-1alpha functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol. Metab. 2018, 7, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sun, C.; Zhou, Y.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef]
- Park, S.W.; Herrema, H.; Salazar, M.; Cakir, I.; Cabi, S.; Basibuyuk Sahin, F.; Chiu, Y.H.; Cantley, L.C.; Ozcan, U. BRD7 regulates XBP1s’ activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 2014, 20, 73–84. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; Ozcan, U. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef]
- Liu, J.; Ibi, D.; Taniguchi, K.; Lee, J.; Herrema, H.; Akosman, B.; Mucka, P.; Salazar Hernandez, M.A.; Uyar, M.F.; Park, S.W.; et al. Inflammation Improves Glucose Homeostasis through IKKbeta-XBP1s Interaction. Cell 2016, 167, 1052–1066. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Ma, K.; Vattem, K.M.; Wek, R.C. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 2002, 277, 18728–18735. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Cavener, D.R. Translational control and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2357–2371. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Teuber, V.; Albert-Gasco, H.; Auyeung, V.C.; Papa, F.R.; Mallucci, G.R.; Hetz, C. Small Molecules to Improve ER Proteostasis in Disease. Trends Pharmacol. Sci. 2019, 40, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Nikesitch, N.; Lee, J.M.; Ling, S.; Roberts, T.L. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, e1007. [Google Scholar] [CrossRef] [Green Version]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef]
- Youle, R.J. Mitochondria-Striking a balance between host and endosymbiont. Science 2019, 365, eaaw9855. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 2015, 350, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Hudson, G. The Ageing Brain, Mitochondria and Neurodegeneration. In Mitochondrial Dysfunction in Neurodegenerative Disorders; Reeve, A.K., Simcox, E.M., Duchen, M.R., Turnbull, D.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 59–80. [Google Scholar]
- Cho, B.; Kim, T.; Huh, Y.J.; Lee, J.; Lee, Y.I. Amelioration of Mitochondrial Quality Control and Proteostasis by Natural Compounds in Parkinson’s Disease Models. Int. J. Mol. Sci. 2019, 20, 5208. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; del Rey, N.L.-G. Parkinson’s Disease-Associated Mutations Affect Mitochondrial Function. In Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease; Buhlman, L.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 139–158. [Google Scholar]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first-in-class cancer metabolism drug. Nat. Rev. Drug Discov. 2017, 16, 593. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Tallman, M.S.; de Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, A.R.; Entenberg, D.; Wang, Y.; Condeelis, J.; Aguirre-Ghiso, J.A. The Different Routes to Metastasis via Hypoxia-Regulated Programs. Trends Cell Biol. 2018, 28, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, K.S.; McNeill, L.A.; Elkins, J.M.; Schofield, C.J. The role of iron and 2-oxoglutarate oxygenases in signalling. Biochem. Soc. Trans. 2003, 31, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef]
- Lee, K.E.; Simon, M.C. SnapShot: Hypoxia-Inducible Factors. Cell 2015, 163, 1288. [Google Scholar] [CrossRef]
- Thompson, C.B. Into Thin Air: How We Sense and Respond to Hypoxia. Cell 2016, 167, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Ast, T.; Meisel, J.D.; Patra, S.; Wang, H.; Grange, R.M.H.; Kim, S.H.; Calvo, S.E.; Orefice, L.L.; Nagashima, F.; Ichinose, F.; et al. Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell 2019, 177, 1507–1521. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Cheng, K.; Ho, K.; Stokes, R.; Scott, C.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Loudovaris, T.; Kay, T.W.; Kulkarni, R.N.; et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J. Clin. Investig. 2010, 120, 2171–2183. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C.W.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2alpha-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat. Med. 2013, 19, 1331–1337. [Google Scholar] [CrossRef]
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chiang, H.H.; Louw, M.; Susanto, A.; Chen, D. Nutrient Sensing and the Oxidative Stress Response. Trends Endocrinol. Metab. 2017, 28, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupre-Crochet, S.; Erard, M.; Nubetae, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Mullebner, A.; Dorighello, G.G.; Kozlov, A.V.; Duvigneau, J.C. Interaction between Mitochondrial Reactive Oxygen Species, Heme Oxygenase, and Nitric Oxide Synthase Stimulates Phagocytosis in Macrophages. Front. Med. 2017, 4, 252. [Google Scholar] [CrossRef]
- Iovine, N.M.; Pursnani, S.; Voldman, A.; Wasserman, G.; Blaser, M.J.; Weinrauch, Y. Reactive nitrogen species contribute to innate host defense against Campylobacter jejuni. Infect. Immun. 2008, 76, 986–993. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Carruba, M.O. Nitric oxide and mitochondrial biogenesis. J. Cell Sci. 2006, 119, 2855–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Campbell, A.; Solaimani, P. Oxidative and Inflammatory Pathways in Age-Related Chronic Disease Processes. In Inflammation, Aging, and Oxidative Stress; Bondy, S.C., Campbell, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 95–106. [Google Scholar]
- Dasgupta, A.; Klein, K. Antioxidants in Food, Vitamins and Supplements; Elsevier: San Diego, CA, USA, 2014; pp. 129–150. [Google Scholar]
- Jeong, Y.; Hellyer, J.A.; Stehr, H.; Hoang, N.T.; Niu, X.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Wakelee, H.A.; et al. Role of KEAP1/NFE2L2 mutations in the chemotherapeutic response of non-small cell lung cancer patients. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, P.P.; Guo, H.T.; Liu, Y.; Li, J.; He, X.J.; Lu, A.P. Safety Profiles of Tripterygium wilfordii Hook F: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2016, 7, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Liu, J.; Feng, X.; Salazar Hernandez, M.A.; Mucka, P.; Ibi, D.; Choi, J.W.; Ozcan, U. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat. Med. 2016, 22, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirjalili, M.H.; Moyano, E.; Bonfill, M.; Cusido, R.M.; Palazon, J. Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules 2009, 14, 2373–2393. [Google Scholar] [CrossRef] [PubMed]
- Winters, M. Ancient medicine, modern use: Withania somnifera and its potential role in integrative oncology. Altern. Med. Rev. 2006, 11, 269–277. [Google Scholar] [PubMed]
- Mishra, L.C.; Singh, B.B.; Dagenais, S. Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): A review. Altern. Med. Rev. 2000, 5, 334–346. [Google Scholar] [PubMed]
- Zhang, M.; Luo, H.; Xi, Z.; Rogaeva, E. Drug repositioning for diabetes based on ‘omics’ data mining. PLoS ONE 2015, 10, e0126082. [Google Scholar] [CrossRef]
- Dyle, M.C.; Ebert, S.M.; Cook, D.P.; Kunkel, S.D.; Fox, D.K.; Bongers, K.S.; Bullard, S.A.; Dierdorff, J.M.; Adams, C.M. Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy. J. Biol. Chem. 2014, 289, 14913–14924. [Google Scholar] [CrossRef]
- Farooq, F.; Balabanian, S.; Liu, X.; Holcik, M.; MacKenzie, A. p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum. Mol. Genet. 2009, 18, 4035–4045. [Google Scholar] [CrossRef] [Green Version]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3, 96ra76. [Google Scholar] [CrossRef]
- Vanderstocken, G.; Dvorkin-Gheva, A.; Shen, P.; Brandsma, C.A.; Obeidat, M.; Bosse, Y.; Hassell, J.A.; Stampfli, M.R. Identification of Drug Candidates to Suppress Cigarette Smoke-induced Inflammation via Connectivity Map Analyses. Am. J. Respir. Cell Mol. Biol. 2018, 58, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dai, Z.; Luo, Z.; Zuo, C. Identification of Pyrvinium, an Anthelmintic Drug, as a Novel Anti-Adipogenic Compound Based on the Gene Expression Microarray and Connectivity Map. Molecules 2019, 24, 2391. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Batkai, S.; Patel, V.; Saito, K.; Matsumoto, S.; Kashiwaya, Y.; Horvath, B.; Mukhopadhyay, B.; Becker, L.; et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, A.L.; Reierson, G.W.; Smith, R.P.; Goldberg, J.L.; Lemmon, V.P.; Bixby, J.L. A chemical genetic approach identifies piperazine antipsychotics as promoters of CNS neurite growth on inhibitory substrates. Mol. Cell. Neurosci. 2012, 50, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.R.; Joshi, S.; Zulcic, M.; Alcaraz, M.; Garlich, J.R.; Morales, G.A.; Cho, Y.J.; Bao, L.; Levy, M.L.; Newbury, R.; et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS ONE 2016, 11, e0150836. [Google Scholar] [CrossRef] [PubMed]
- Muthuswami, M.; Ramesh, V.; Banerjee, S.; Viveka Thangaraj, S.; Periasamy, J.; Bhaskar Rao, D.; Barnabas, G.D.; Raghavan, S.; Ganesan, K. Breast tumors with elevated expression of 1q candidate genes confer poor clinical outcome and sensitivity to Ras/PI3K inhibition. PLoS ONE 2013, 8, e77553. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Xiang, B.; Guix, M.; Olivares, M.G.; Parker, J.; Chung, C.H.; Pandiella, A.; Arteaga, C.L. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol. Cell. Biol. 2008, 28, 5605–5620. [Google Scholar] [CrossRef]
- Wang, G.; Ye, Y.; Yang, X.; Liao, H.; Zhao, C.; Liang, S. Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma. PLoS ONE 2011, 6, e14573. [Google Scholar] [CrossRef]
- Fortney, K.; Griesman, J.; Kotlyar, M.; Pastrello, C.; Angeli, M.; Sound-Tsao, M.; Jurisica, I. Prioritizing therapeutics for lung cancer: An integrative meta-analysis of cancer gene signatures and chemogenomic data. PLoS Comput. Biol. 2015, 11, e1004068. [Google Scholar] [CrossRef]
- Cheng, H.W.; Liang, Y.H.; Kuo, Y.L.; Chuu, C.P.; Lin, C.Y.; Lee, M.H.; Wu, A.T.; Yeh, C.T.; Chen, E.I.; Whang-Peng, J.; et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015, 6, e1753. [Google Scholar] [CrossRef]
- Rho, S.B.; Kim, B.R.; Kang, S. A gene signature-based approach identifies thioridazine as an inhibitor of phosphatidylinositol-3’-kinase (PI3K)/AKT pathway in ovarian cancer cells. Gynecol. Oncol. 2011, 120, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.; Stegmaier, K.; Raj, S.M.; et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedemann, R.E.; Schmidt, J.; Keats, J.J.; Shi, C.X.; Zhu, Y.X.; Palmer, S.E.; Mao, X.; Schimmer, A.D.; Stewart, A.K. Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-kappaB with antimyeloma activity in vitro and in vivo. Blood 2009, 113, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Zador, Z.; King, A.T.; Geifman, N. New drug candidates for treatment of atypical meningiomas: An integrated approach using gene expression signatures for drug repurposing. PLoS ONE 2018, 13, e0194701. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Li, X.; Gutierrez, A.; Ahn, Y.; Neuberg, D.S.; O’Neil, J.; Strack, P.R.; Winter, C.G.; Winter, S.S.; Larson, R.S.; et al. Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood 2010, 115, 1735–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassane, D.C.; Guzman, M.L.; Corbett, C.; Li, X.; Abboud, R.; Young, F.; Liesveld, J.L.; Carroll, M.; Jordan, C.T. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 2008, 111, 5654–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, S.A.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Belver, L.; Xu, L.; Qin, Y.; Kageyama, R.; Ferrando, A.A. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood 2015, 125, 2806–2814. [Google Scholar] [CrossRef] [Green Version]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.C.; et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef]
- Rosenbluth, J.M.; Mays, D.J.; Pino, M.F.; Tang, L.J.; Pietenpol, J.A. A gene signature-based approach identifies mTOR as a regulator of p73. Mol. Cell. Biol. 2008, 28, 5951–5964. [Google Scholar] [CrossRef]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef]
- Stockwell, S.R.; Platt, G.; Barrie, S.E.; Zoumpoulidou, G.; Te Poele, R.H.; Aherne, G.W.; Wilson, S.C.; Sheldrake, P.; McDonald, E.; Venet, M.; et al. Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS ONE 2012, 7, e28568. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Kim, S.; Lee, Y.-I.; Lee, J. Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap). Int. J. Mol. Sci. 2019, 20, 5601. https://doi.org/10.3390/ijms20225601
Gao Y, Kim S, Lee Y-I, Lee J. Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap). International Journal of Molecular Sciences. 2019; 20(22):5601. https://doi.org/10.3390/ijms20225601
Chicago/Turabian StyleGao, Yurong, Sungwoo Kim, Yun-Il Lee, and Jaemin Lee. 2019. "Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap)" International Journal of Molecular Sciences 20, no. 22: 5601. https://doi.org/10.3390/ijms20225601