CRISPR/Cas9-Mediated Knock-Out of KrasG12D Mutated Pancreatic Cancer Cell Lines

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
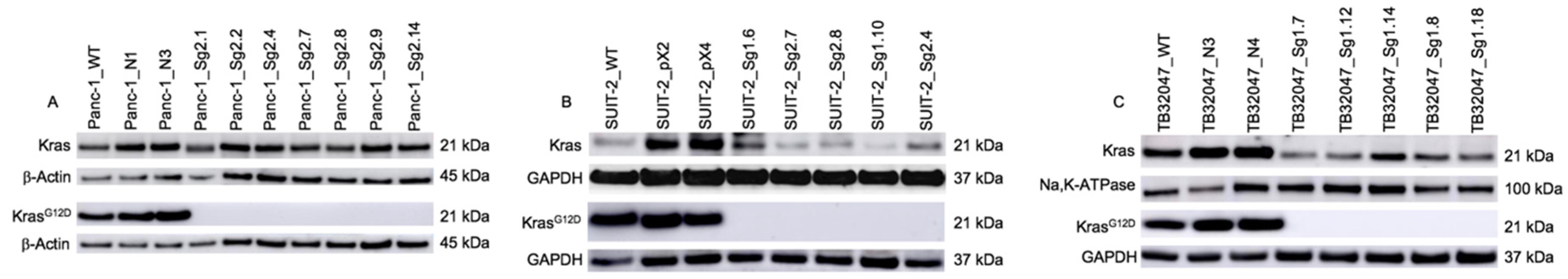
2.1. Expression of KrasG12D and Total Ras after CRISPR/Cas9-Mediated Knock-Out in PaCa Cell Lines
2.2. DNA Sequencing from Knocked-Out Clones
2.3. Expression of Key Signal Transduction Proteins in CRISPR/Cas9-Edited PaCa Cell Lines
2.4. TB32047 RNA Sequencing Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRISPR/Cas9 Gene Editing
4.3. Western Blot
4.4. Confirmation of CRISPR/Cas9-Mediated Knock-out
4.5. RNA Isolation and Preparation
4.6. RNA Sequencing
4.7. Functional and Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| bp | Base pair |
| Cas | CRISPR-associated |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| DEG(s) | Differentially expressed gene(s) |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EDTA | Ethylenediaminetetraacetic acid |
| FBS | Fetal bovine serum |
| Indel | Inserts/deletions |
| MEM | Minimum essential media |
| NC | Negative control |
| PaCa | Pancreatic cancer |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI3K | Phosphatidyl inositol 3-kinase |
| RIPA | Radioimmunoprecipitation assay |
| RPMI | Roswell Park Memorial Institute |
| seq | Sequencing |
| WB | Western blot |
| WT | Wildtype |
References
- American Cancer Society. Cancer Facts & Figures 2018; American Cancer Society: Atlanta, GA, USA, 2018; pp. 4–8. [Google Scholar]
- Krebsdaten.de. Available online: https://www.krebsdaten.de/Krebs/DE/Content/Krebsarten/Bauchspeicheldruesenkrebs/bauchspeicheldruesenkrebs.html (accessed on 13 March 2019).
- Larsson, S.C.; Permet, J.; Håkansson, N.; Näslund, I.; Bergkvist, L.; Wolk, A. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. Br. J. Cancer 2005, 93, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, U.; Riess, H. Current standards in the treatment of pancreatic cancer. Dtsch. Med. Wochenschr. 2015, 140, 1813–1816. [Google Scholar] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Rassow, J.; Hauser, K.; Netzker, R.; Deutzmann, R. Duale Reihe: Biochemie, 3rd ed.; Thieme: Stuttgart, Germany, 2012; ISBN 978-3-13-125353-8. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef]
- Chen, P.Y.; Muzumdar, M.D.; Dorans, K.J.; Robbins, R.; Bhutkar, A.; del Rosario, A.; Mertins, P.; Qiao, J.; Schafer, A.C.; Gertler, F.; et al. Adaptive and Reversible Resistance to Kras Inhibition in Pancreatic Cancer Cells. Cancer Res. 2018, 78, 985–1002. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef]
- Charpentier, E.; Doudna, J.A. Biotechnology: Rewriting a genome. Nature 2013, 495, 50–51. [Google Scholar] [CrossRef]
- Hale, C.R.; Zhao, P.; Olson, S.; Duff, M.O.; Graveley, B.R.; Wells, L.; Terns, R.M.; Terns, M.P. RNA guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 2009, 139, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual- RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F., III; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Hirano, T.; Katsuhiko Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef]
- Petz, M.; Them, N.; Huber, H.; Beug, H.; Mikulits, W. La enhances IRES-mediated translation of laminin B1 during malignant epithelial to mesenchymal transition. Nucleic Acids Res. 2012, 40, 290–302. [Google Scholar] [CrossRef]
- Anderson, C.M.; Andreas Stahl, A. SLC27 fatty acid transport proteins. Mol. Aspects Med. 2013, 34, 516–528. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.J.; Yuen, G.; Luo, J. Multiplexed CRISPR/Cas9 gene knockout with simple crRNA: tracrRNA co-transfection. Cell Biosci. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 October 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 20, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2017, 46, 754–761. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Panc-1 Clones | Indel Size | SUIT-2 Clones | Indel Size | TB32047 Clones | Indel Size |
|---|---|---|---|---|---|
| 2.1 | small | 1.6 | small | 1.7 | small |
| 2.2 | small | 2.7 | small | 1.12 | small |
| 2.4 | large | 2.8 | small | 1.14 | small |
| 2.7 | single nucleotide | 1.10 | small | 1.8 | large |
| 2.8 | small | 2.4 | large | 1.18 | small |
| 2.9 | small | ||||
| 2.14 | single nucleotide |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lentsch, E.; Li, L.; Pfeffer, S.; Ekici, A.B.; Taher, L.; Pilarsky, C.; Grützmann, R. CRISPR/Cas9-Mediated Knock-Out of KrasG12D Mutated Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 5706. https://doi.org/10.3390/ijms20225706
Lentsch E, Li L, Pfeffer S, Ekici AB, Taher L, Pilarsky C, Grützmann R. CRISPR/Cas9-Mediated Knock-Out of KrasG12D Mutated Pancreatic Cancer Cell Lines. International Journal of Molecular Sciences. 2019; 20(22):5706. https://doi.org/10.3390/ijms20225706
Chicago/Turabian StyleLentsch, Eva, Lifei Li, Susanne Pfeffer, Arif B. Ekici, Leila Taher, Christian Pilarsky, and Robert Grützmann. 2019. "CRISPR/Cas9-Mediated Knock-Out of KrasG12D Mutated Pancreatic Cancer Cell Lines" International Journal of Molecular Sciences 20, no. 22: 5706. https://doi.org/10.3390/ijms20225706