Impact of Glucoraphanin-Mediated Activation of Nrf2 on Non-Alcoholic Fatty Liver Disease with a Focus on Mitochondrial Dysfunction


{kind=link}
{kind=link}
Abstract
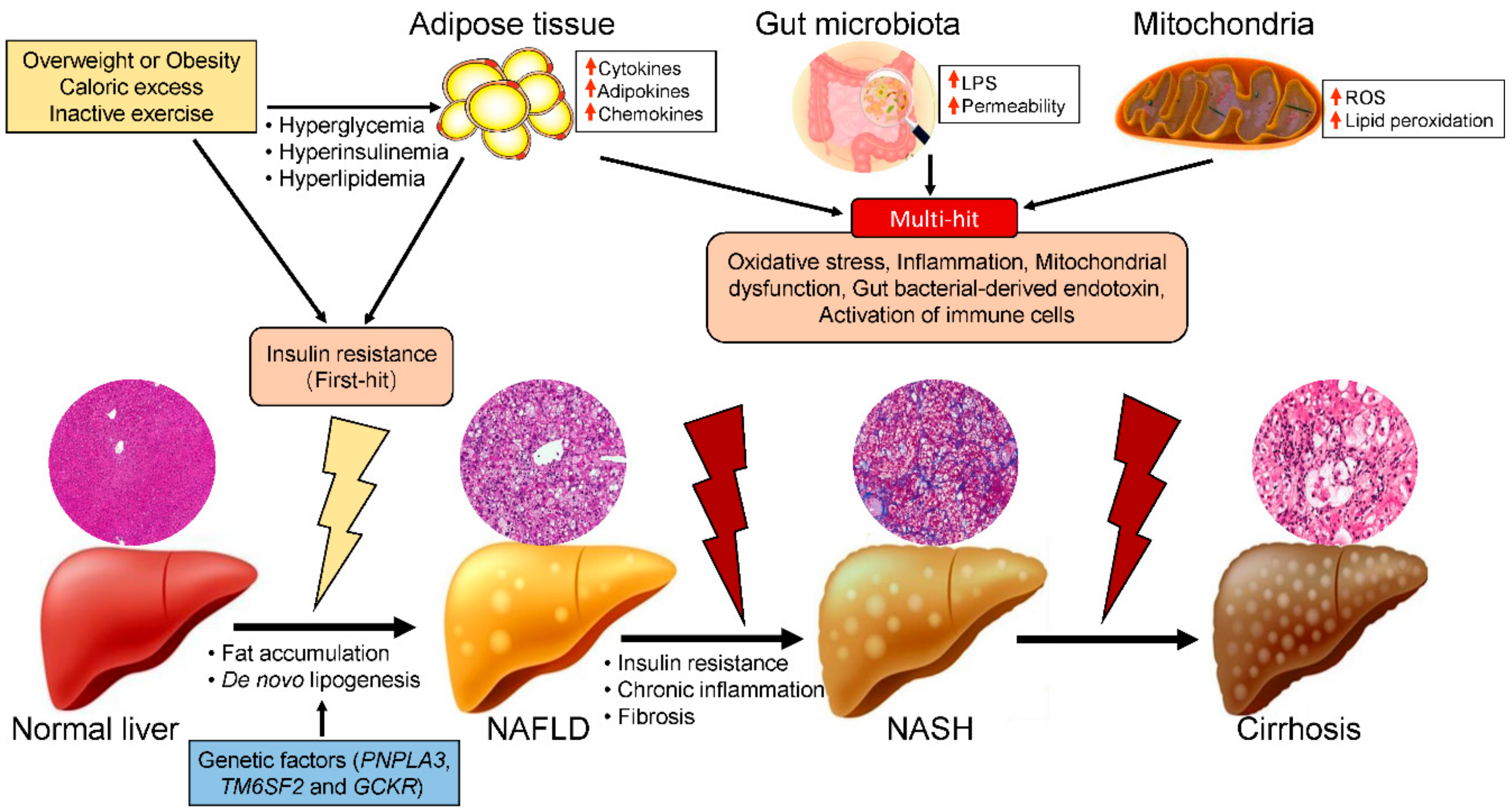
:1. Introduction
2. Risk Factors for NASH
3. Roles of Mitochondrial Dysfunction in the Pathogenesis of NASH
4. The Nrf2-Keap1 Pathway in NASH
5. Impact of Glucoraphanin on Mitochondrial Dysfunction-Related NASH
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| ROS | Reactive oxygen species |
| Nrf | Nuclear factor erythroid 2-related factor |
| TG | Triglycerides |
| IL | Interleukin |
| MCP | Monocyte chemoattractant protein |
| TNF | Tumor necrosis factor |
| FFA | Free fatty acid |
| LPS | Lipopolysaccharide |
| IFN | Interferon |
| ATM | Adipose tissue macrophage |
| KC | Kupffer cell |
| PNPLA3 | Patatin-like phospholipase domain-containing 3 |
| TM6SF2 | Transmembrane 6 superfamily 2 |
| GCKR | Glucokinase regulatory protein |
| SNP | Single-nucleotide polymorphism |
| ATP | Adenosine triphosphate |
| CPT | Carnitine palmitoyl transferase |
| SREBP | Sterol regulatory element-binding protein |
| PI3K | Phosphoinositide 3-kinase |
| LXR | Liver X receptor |
| FAS | Fatty-acid synthase |
| ACC | Acetyl CoA carboxylase |
| SCD | Stearoyl-CoA desaturase |
| ChREBP | Carbohydrate-responsive element-binding protein |
| VLDL | Very low-density lipoprotein |
| AMPK | AMP-activated protein kinase |
| PPAR | Peroxisome proliferator-activated receptor |
| mtDNA | Mitochondrial DNA |
| Keap1 | kelch-like ECH-associated protein 1 |
| HFD | High-fat diet |
| NOX | NADPH oxidase |
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Loomba, R.; Anstee, Q.M.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology 2018, 68, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Makhlouf, H.R. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef]
- Araujo, A.R.; Rosso, N.; Bedogni, G.; Tiribelli, C.; Bellentani, S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: What we need in the future. Liver Int. 2018, 38 (Suppl. 1), 47–51. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Yu, E.L.; Golshan, S.; Harlow, K.E.; Angeles, J.E.; Durelle, J.; Goyal, N.P.; Newton, K.P.; Sawh, M.C.; Hooker, J.; Sy, E.Z.; et al. Prevalence of nonalcoholic fatty liver disease in children with obesity. J. Pediatr. 2019, 207, 64–70. [Google Scholar] [CrossRef]
- Rivera-Andrade, A.; Kroker-Lobos, M.F.; Lazo, M.; Freedman, N.D.; Smith, J.W.; Torres, O.; McGlynn, K.A.; Groopman, J.D.; Guallar, E.; Ramirez-Zea, M. High prevalence of non-alcoholic fatty liver disease and metabolic risk factors in Guatemala: A population-based study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 191–200. [Google Scholar] [CrossRef]
- Argo, C.K.; Caldwell, S.H. Epidemiology and natural history of non-alcoholic steatohepatitis. Clin. Liver Dis. 2009, 13, 511–531. [Google Scholar] [CrossRef]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. Hepatic steatosis: Innocent bystander or guilty party. Hepatology 1998, 27, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.J.O. Steatohepatitis: A tale of two “hits”. Gastroenterology 1998, 114, 842–885. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Koppe, S.W. Obesity and the liver: Nonalcoholic fatty liver disease. Transl. Res. 2014, 164, 312–322. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharm. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef]
- Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules 2015, 5, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Mustafa, G.; Alam, M.; Ahmad, N. Insulin resistance in development and progression of nonalcoholic fatty liver disease. World J. Gastrointest. Pathophysiol. 2016, 7, 211. [Google Scholar] [CrossRef]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front. Endocrinol. 2012, 3, 181. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflug. Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 2017, 20, 137–149. [Google Scholar]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.J.; Sadygov, R.G.; et al. Hepatic mitochondrial defects in a mouse model of NAFLD are associated with increased degradation of oxidative phosphorylation subunits. Mol. Cell. Proteom. 2018, 17, 2371–2386. [Google Scholar]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell. Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Lazo, M.; Solga, S.F.; Horska, A.; Bonekamp, S.; Diehl, A.M.; Brancati, F.L.; Wagenknecht, L.E.; Pi-Sunyer, F.X.; Kahn, S.E.; Clark, J.M.; et al. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care 2010, 33, 2156–2163. [Google Scholar] [CrossRef] [Green Version]
- Mancina, R.M.; Matikainen, N.; Maglio, C.; Soderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef] [Green Version]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.A.; Larrieta, E.; Calva, J.J.; Kershenobich, D.; Torre, A. The expression of PNPLA3 polymorphism could be the key for severe liver disease in NAFLD in hispanic population. Ann. Hepatol. 2017, 16, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Byoun, Y.S.; Jeong, S.H.; Woo, B.H.; Jang, E.S.; Kim, J.W.; Kim, H.Y. Role of the PNPLA3 I148M polymorphism in nonalcoholic fatty liver disease and fibrosis in Korea. Dig. Dis. Sci. 2014, 59, 2967–2974. [Google Scholar] [CrossRef] [PubMed]
- Okanoue, T.; Kawaguchi, T.; Sumida, Y.; Umemura, A.; Matsuo, K.; Takahashi, M.; Yasui, K.; Saibara, T.; Hashimoto, E.; Kawanaka, M.; et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS ONE 2012, 7, e38322. [Google Scholar]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyotylainen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursier, J.M.O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; Hunault, G.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Derm. 2013, 169 (Suppl. 2), 1–8. [Google Scholar]
- Nicolson, G.L. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr. Med. Clin. J. 2014, 13, 35. [Google Scholar]
- Oliveira, C.P.C.A.; Barbeiro, H.V.; Lima, V.M.; Soriano, F.; Ribeiro, C.; Molan, N.A.; Alves, V.A.; Souza, H.P.; Machado, M.C.; Carrilho, F.J. Liver mitochondrial dysfunction and oxidative stress in the pathogenesis of experimental nonalcoholic fatty liver disease. Braz. J. Med. Biol. Res. 2006, 39, 189–194. [Google Scholar]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084. [Google Scholar] [CrossRef]
- Paglialunga, S.; Dehn, C.A. Clinical assessment of hepatic de novo lipogenesis in non-alcoholic fatty liver disease. Lipids Health Dis. 2016, 15, 159. [Google Scholar] [CrossRef] [Green Version]
- Guillet-Deniau, I.M.V.; Le Lay, S.; Achouri, Y.; Carré, D.; Girard, J.; Foufelle, F.; Ferré, P. Sterol regulatory element binding protein-1c expression and action in rat muscles: Insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes Care 2002, 51, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Transcriptional mediators of lipid homeostasis. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 491–498. [Google Scholar]
- Zhang, D.; Tong, X.; VanDommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, M.B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867. [Google Scholar] [CrossRef] [Green Version]
- Mcgarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.G.; Robson, S.C.; Yao, Z. Lipoprotein metabolism in nonalcoholic fatty liver disease. J. Biomed. Res. 2013, 27, 1. [Google Scholar] [PubMed] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMPK: Sensing energy while talking to other signaling pathways. Cell Metab. 2014, 20, 939–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK: Mediated regulation of lipid metabolism by phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Fuentes, E.; Guzman-Jofre, L.; Moore-Carrasco, R.; Palomo, I. Role of PPARs in inflammatory processes associated with metabolic syndrome (Review). Mol. Med. Rep. 2013, 8, 1611–1616. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zheng, F.; Pan, Q.; Zhang, S.; Yu, D.; Xu, Z.; Li, H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J. Mol. Endocrinol. 2015, 55, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodoro, J.S.; Rolo, A.P.; Duarte, F.V.; Simoes, A.M.; Palmeira, C.M. Differential alterations in mitochondrial function induced by a choline-deficient diet: Understanding fatty liver disease progression. Mitochondrion 2008, 8, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Kamei, J.; Ishikawa, H.; Komatsu, Y.; Kaneko, S.; et al. A porcine placental extract prevents steatohepatitis by suppressing activation of macrophages and stellate cells in mice. Oncotarget 2018, 9, 15047–15060. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1349. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.K.J.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharm. 2010, 245, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, M.; Ushida, Y.; Shiozawa, H.; Umeda, R.; Tsuruya, K.; Aoki, Y.; Suganuma, H.; Nishizaki, Y. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J. Gastroenterol. 2015, 21, 12457–12467. [Google Scholar] [CrossRef] [PubMed]
- Glade, M.J.; Meguid, M.M. A Glance at... Broccoli, glucoraphanin, and sulforaphane. Nutrition 2015, 31, 1175–1178. [Google Scholar] [CrossRef]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.L.; Jacobson, L.P.; Munoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef]
- Seo, H.A.; Lee, I.K. The role of Nrf2: Adipocyte differentiation, obesity, and insulin resistance. Oxid. Med. Cell. Longev. 2013, 2013, 184598. [Google Scholar] [CrossRef] [Green Version]
- Meher, A.K.; Sharma, P.R.; Lira, V.A.; Yamamoto, M.; Kensler, T.W.; Yan, Z.; Leitinger, N. Nrf2 deficiency in myeloid cells is not sufficient to protect mice from high-fat diet-induced adipose tissue inflammation and insulin resistance. Free Radic. Biol. Med. 2012, 52, 1708–1715. [Google Scholar] [CrossRef] [Green Version]
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin Ameliorates Obesity and Insulin Resistance Through Adipose Tissue Browning and Reduction of Metabolic Endotoxemia in Mice. Diabetes 2017, 66, 1222–1236. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mealey, G.B.; Berry, W.L.; Plafker, S.M. Sulforaphane is a Nrf2-independent inhibitor of mitochondrial fission. Redox Biol. 2017, 11, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Pozo, C.; Tan, K.N.; Gotteland, M.; Borges, K. Sulforaphane protects against high cholesterol-induced mitochondrial bioenergetics impairments, inflammation, and oxidative stress and preserves pancreatic beta-cells function. Oxid. Med. Cell. Longev. 2017, 2017, 3839756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Pozo, C.; Tan, K.N.; Borges, K. Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 2015, 135, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Armah, C.N.; Traka, M.H.; Dainty, J.R.; Defernez, M.; Janssens, A.; Leung, W.; Doleman, J.F.; Potter, J.F.; Mithen, R.F. A diet rich in high-glucoraphanin broccoli interacts with genotype to reduce discordance in plasma metabolite profiles by modulating mitochondrial function. Am. J. Clin. Nutr. 2013, 98, 712–722. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Nagata, N.; Ota, T. Impact of Glucoraphanin-Mediated Activation of Nrf2 on Non-Alcoholic Fatty Liver Disease with a Focus on Mitochondrial Dysfunction. Int. J. Mol. Sci. 2019, 20, 5920. https://doi.org/10.3390/ijms20235920
Xu L, Nagata N, Ota T. Impact of Glucoraphanin-Mediated Activation of Nrf2 on Non-Alcoholic Fatty Liver Disease with a Focus on Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2019; 20(23):5920. https://doi.org/10.3390/ijms20235920
Chicago/Turabian StyleXu, Liang, Naoto Nagata, and Tsuguhito Ota. 2019. "Impact of Glucoraphanin-Mediated Activation of Nrf2 on Non-Alcoholic Fatty Liver Disease with a Focus on Mitochondrial Dysfunction" International Journal of Molecular Sciences 20, no. 23: 5920. https://doi.org/10.3390/ijms20235920