The 1,10-Phenanthroline Ligand Enhances the Antiproliferative Activity of DNA-Intercalating Thiourea-Pd(II) and -Pt(II) Complexes Against Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cell Lines

 , , , , , and
, , , , , and
Abstract
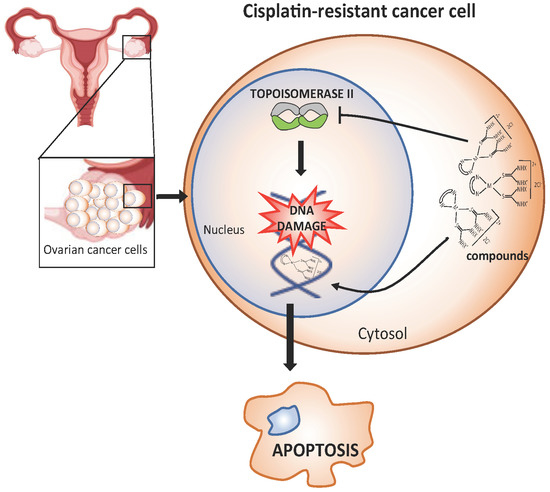
:
1. Introduction
2. Results
2.1. Comparison of the Cytotoxic Efficacy of the Different Complexes
2.2. Remarkable Intracellular Accumulation of the Pd(phen) Complexes
2.3. Pd(phen) Complexes Showed the Highest Affinity for DNA and Intercalation Ability
2.4. Pd(phen) Complexes Caused the Greatest Perturbation of Cell Cycle and Apoptosis Induction
2.5. Pd(phen) Complexes Targeted TOPO-II and Thymidylate Synthase (TS) Activity Affecting DNA Integrity
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Cell Lines
4.3. Cell Growth Assay
4.4. Intracellular Complex Accumulation
4.5. Measurement of DNA-Intercalating Ability
4.6. TOPO-II Decatenation Assay
4.7. Flow Cytometric Analysis of Cell Cycle
4.8. Western Immunoblotting
4.9. TS Catalytic Assay
4.10. Radioligand Assay for Folic Acid
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cDDP | Cisplatin or cis-diamminedichloridoplatinum(II) |
| Phen | Phenanthroline |
| Bipy | Bipyridine |
| Tu | Thiourea |
| TOPO-II | Topoisomerase-II |
| TS | Thymidylate synthase |
| DHFR | Dihydrofolate reductase |
| ICP-MS | Inductively-coupled plasma mass spectrometry |
| EB | Ethidium bromide |
| FA | Folic acid |
| P-gp | P-glycoprotein |
References
- Topotecan, Pegylated Liposomal Doxorubicin Hydrochloride, Paclitaxel, Trabectedin and Gemcitabine for Treating Recurrent Ovarian Cancer; National Institute for Health and Care Excellence (NICE): London, UK, 2016.
- Neto, B.A.D.; Lapis, A.A.M. Recent Developments in the Chemistry of Deoxyribonucleic Acid (DNA) Intercalators: Principles, Design, Synthesis, Applications and Trends. Molecules 2009, 14, 1725–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Nicolò, F.; Nordén, B.; Lincoln, P. Ambivalent intercalators for DNA: L-shaped platinum(II) complexes. Inorg. Chem. 2004, 43, 2416–2421. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Vainiglia, P.A. The intercalation to DNA of bipyridyl complexes of platinum(II) with thioureas. J. Inorg. Biochem. 2005, 99, 560–565. [Google Scholar] [CrossRef]
- Marverti, G.; Cusumano, M.; Ligabue, A.; Di Pietro, M.L.; Vainiglia, P.; Ferrari, A.; Bergomi, M.; Moruzzi, M.S.; Frassineti, C. Studies on the antiproliferative effects of novel DNA-intercalating bypiridylthiourea-Pt(II) complexes against cisplatin-sensitive and -resistant human ovarian cancer cells. J. Inorg. Biochem. 2008, 102, 699–712. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwoun, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Muggia, F. Platinum compounds 30 years after the introduction of cisplatin: Implications for the treatment of ovarian cancer. Gynecol. Oncol. 2009, 112, 275–281. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Montanari, M.; Guerrieri, D.; Cusumano, M.; Di Pietro, M.L.; Troiano, L.; Di Vono, E.; Iotti, S.; Farruggia, G.; et al. Characterization of the cell growth inhibitory effects of a novel DNA-intercalating bipyridyl-thiourea-Pt(II) complex in cisplatin-sensitive and –resistant human ovarian cancer cells. Investig. New Drugs 2011, 29, 73–81. [Google Scholar] [CrossRef]
- Rotondo, A.; Barresi, S.; Cusumano, M.; Rotondo, E. Structural and dynamic NMR characterization of [Pd(bipy)(R-thiourea)2]2+ and [Pd(phen)(R-thiourea)2] 2+ cations. Polyhedron 2012, 45, 23–29. [Google Scholar] [CrossRef]
- Rotondo, A.; Barresi, S.; Cusumano, M.; Rotondo, E.; Donato, P.; Mondello, L. NMR characterisation and dynamic behaviour of [Pt(bipy) (R-Thiourea)2]Cl2 and [Pt(phen)(R-Thiourea)2]Cl2 complexes. Inorg. Chim. Acta 2014, 410, 1–10. [Google Scholar] [CrossRef]
- Hazarika, P.; Bezbaruah, B.; Das, P.; Medhi, O.K.; Medhi, C. A model study on the stacking interaction of phenanthroline ligand with nucleic acid base pairs: An ab initio, MP2 and DFT studies. J. Inorg. Biochem. 2011, 2, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.V.; Sakore, T.D.; Sobell, H.M. Structure of a novel drug-nucleic acid crystalline com-plex: 1,10-phenanthroline-platinum (II) ethylenedia-mine-5′-phosphoryl-thymidylyl (3′-5′) deoxyadenosine. J. Biomol. Struct. Dyn. 1984, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Barra, C.V.; Rocha, F.V.; Morel, L.; Gautier, A.; Garrido, S.S.; Mauro, A.E.; Frem, R.C.G.; Netto, A.V.G. DNA binding, topoisomerase inhibition and cytotoxicity of palladium(II) complexes with 1,10-phenanthroline and thioureas. Inorg. Chim. Acta 2016, 446, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Kemp, S.; Wheate, N.J.; Buck, D.P.; Nikac, M.; Collins, J.G.; Aldrich-Wright, J.R. The effect of ancillary ligand chirality and phenanthroline functional group substitution on the cytotoxicity of platinum(II)-based metallointercalators. J. Inorg. Biochem. 2007, 101, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; De Dille, A.; Jameson, V.J.; Smith, L.; Dernell, W.S.; Manning, M.C. Improved potency of cisplatin by hydrophobic ion pairing. Cancer Chemother. Pharmacol. 2004, 54, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A. DNA Interaction of Platinum(II) Complexes with 1,10-Phenanthroline and Extended Phenanthrolines. Inorg. Chem. 2006, 45, 1230–1235. [Google Scholar] [CrossRef]
- Geall, A.J.; Blagbrough, I.S. Rapid and sensitive ethidium bromide fluorescence quenching assay of polyamine conjugate-DNA interactions for the analysis of lipoplex formation in gene therapy. J. Pharm. Biomed. Anal. 2000, 22, 849–859. [Google Scholar] [CrossRef]
- Alonso, A.; Almendral, M.J.; Curto, Y.; Criado, J.J.; Rodrıguez, E.; Manzano, J.L. Determination of the DNA-binding characteristics of ethidium bromide, proflavine, and cisplatin by flow injection analysis: Usefulness in studies on antitumor drugs. Anal. Biochem. 2006, 355, 157–164. [Google Scholar] [CrossRef]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase-II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Nishiyama, M.; Yamamoto, W.; Park, J.S.; Okamoto, R.; Hanaoka, H.; Takano, H.; Saito, N.; Matsukawa, M.; Shirasaka, T.; Kurihara, M. Low-Dose Cisplatin and 5-Fluorouracil in Combination Can Repress Increased Gene Expression of Cellular Resistance Determinants to Themselves. Clin. Cancer Res. 1999, 5, 2620–2628. [Google Scholar]
- Araki, H.; Fukushima, M.; Kamiyama, Y.; Shirasaka, T. Effect of consecutive lower-dose cisplatin in enhancement of 5-Fluorouracil cytotoxicity in experimental tumor cells in vivo. Cancer Lett. 2000, 160, 185–191. [Google Scholar] [CrossRef]
- Morais, T.S.; Valente, A.; Tomaz, A.I.; Marques, F.; Garcia, M.H. Tracking antitumor metallodrugs: Promising agents with the Ru(II)- and Fe(II)-cyclopentadienyl scaffolds. Future Med. Chem. 2016, 8, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Okabe, M.; Shen, D.W.; Liang, X.J.; Gottesman, M.M. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Barnes, K.R.; Lippard, S.J. Cisplatin and Related Anticancer Drugs: Recent Advances and Insights. In Metal Complexes in Tumor Diagnosis and as Anticancer Agents; Sigel, A., Sigel, H., Eds.; Metal Ions in Biolgical Systems; Marcel Dekker: New York, NY, USA, 2004; Volume 42, pp. 143–177. [Google Scholar]
- Liu, H.K.; Sadler, P.J. Metal complexes as DNA intercalators. Acc. Chem. Res. 2011, 44, 349–359. [Google Scholar] [CrossRef]
- Gil, A.; Melle-Franco, G.M.; Branchadell, V.; Calhorda, M.J. How the Intercalation of Phenanthroline Affects the Structure, Energetics, and Bond Properties of DNA Base Pairs: Theoretical Study Applied to Adenine−Thymine and Guanine−Cytosine Tetramers. J. Chem. Theory Comput. 2015, 11, 2714–2728. [Google Scholar] [CrossRef]
- Mišković, K.; Bujak, M.; Baus-Lončar, M.; Glavaš-Obrovac, L. Antineoplastic DNA binding compounds. intercalating and minor groove binding drugs. Arch. Hig Rada Toksikol. 2013, 64, 593–602. [Google Scholar]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gao, F.; Yang, W.Y.; Zhou, Z.X.; Lin, J.Q.; Ji, L.N. Structure-activity relationship of polypyridyl ruthenium(II) complexes as DNA intercalators, DNA photocleavage reagents, and DNA topoisomerase and RNA polymerase inhibitors. Chem. Biodivers. 2013, 10, 367–384. [Google Scholar] [CrossRef]
- Hande, K.R. Topoisomerase II inhibitors. Updat. Cancer Ther. 2006, 1, 3–15. [Google Scholar] [CrossRef]
- Tacara, O.; Sriamornsakb, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharm. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Fornace, A.J., Jr. Mammalian DNA damage-inducible genes associated with growth arrest and apoptosis. Mutat. Res. Rev. Genet. Toxicol. 1996, 340, 109–124. [Google Scholar] [CrossRef]
- Thorna, C.F.; Oshiroa, C.; Marshe, S.; Hernandez-Boussardb, T.; McLeodd, H.; Kleina, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updat. 2015, 23, 20–54. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.H.; Cheng, A.L.; Wan, J.P.; Lin, C.S.; Liu, C.C. Down-regulation of thymidylate synthase expression and its steady-state mRNA by oxaliplatin in colon cancer cell. Anticancer Drugs 2004, 15, 371–376. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Ligasová, A.; Strunin, D.; Friedecký, D.; Adam, T.; Koberna, K. A Fatal Combination: A Thymidylate Synthase Inhibitor with DNA Damaging Activity. PLoS ONE 2015, 10, e0117459. [Google Scholar] [CrossRef]
- Barret, J.M.; Calsou, P.; Larsen, A.K.; Salles, B. A cisplatin-resistant murine leukemia cell line exhibits increased topoisomerase II activity. Mol. Pharmacol. 1994, 46, 431–436. [Google Scholar]
- Minagawa, Y.; Kigawa, J.; Irie, T.; Kanamori, Y.; Itamochi, H.; Cheng, X.; Terakawa, N. Enhanced topoisomerase I activity and increased topoisomerase II alpha content in cisplatin-resistant cancer cell lines. Jpn. J. Cancer Res. 1997, 88, 1218–1223. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Paglietti, G.; Corona, P.; Piras, S.; Vitale, G.; Guerrieri, D.; Luciani, R.; Costi, M.P.; Frassineti, C.; et al. Collateral sensitivity to novel thymidylate synthase inhibitors correlates with folate cycle enzymes impairment in cisplatin-resistant human ovarian cancer cells. Eur. J. Pharmacol. 2009, 615, 17–26. [Google Scholar] [CrossRef] [PubMed]
- DiSaia, P.J.; Morrow, M.; Kanabus, J.; Piechal, W.; Townsend, D.E. Two new tissue culture lines from ovarian cancer. Gynecol. Oncol. 1975, 3, 215–219. [Google Scholar] [CrossRef]
- Marverti, G.; Andrews, P.A. Stimulation of cis-diamminedichloroplatinum(II) accumulation by modulation of passive permeability with genistein: An altered response in accumulation-defective resistant cells. Clin. Cancer Res. 1996, 2, 991–999. [Google Scholar] [PubMed]
- Beaufort, B.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Marverti, G.; Ligabue, A.; Lombardi, P.; Ferrari, S.; Monti, M.G.; Frassineti, C.; Costi, M.P. Modulation of the expression of folate cycle enzymes and polyamine metabolism by berberine in cisplatin-sensitive and -resistant human ovarian cancer cells. Int. J. Oncol. 2013, 43, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Eggera, A.E.; Rappela, C.; Jakupec, M.A.; Hartingera, C.G.; Heffeterb, P.; Keppler, B.K. Development of an experimental protocol for uptake studies of metal compounds in adherent tumor cells. Anal. At. Spectrom. 2009, 24, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.N.; Wang, M.; Zhao, L.C.; Sun, B.Y.; Wang, B.; Chen, H.Q.; Zhao, Y.L.; Chai, Z.F.; Feng, W.Y. Quantitative analysis of Gd@C82(OH)22 and cisplatin uptake in single cells by inductively coupled plasma mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2383–2391. [Google Scholar] [CrossRef]
- Garbett, N.C.; Hammond, N.B.; Graves, D.E. Influence of the Amino Substituents in the Interaction of Ethidium Bromide with DNA. Biophys. J. 2004, 87, 3974–3981. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Di Rocco, G.; Martinelli, I.; Pacifico, S.; Guerrini, R.; Cichero, E.; Fossa, P.; Franchini, S.; Cardarelli, S.; Giorgi, M.; Sola, M.; et al. Fluorometric detection of protein-ligand engagement: The case of phosphodiesterase5. J. Pharm. Biomed. Anal. 2018, 149, 335–342. [Google Scholar] [CrossRef]
- Belluti, S.; Basile, V.; Benatti, P.; Ferrari, E.; Marverti, G.; Imbriano, C. Concurrent inhibition of enzymatic activity and NF-Y-mediated transcription of Topoisomerase-IIα by bis-DemethoxyCurcumin in cancer cells. Cell Death Dis. 2013, 4, e756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolbeare, F.; Gratzner, H.; Pallavicini, M.G.; Gray, J.W. Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc. Natl. Acad. Sci. USA 1983, 80, 5573–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauritz, R.; Peters, G.J.; Kathmann, I.; Teshale, I.; Noordhuis, P.; Comijn, E.M.; Pinedo, H.M.; Jansen, G. Dynamics of antifolate transport via the reduced folate carrier and the membrane folate receptor in murine leukaemia cells in vitro and in vivo. Cancer Chemother. Pharmacol. 2008, 62, 937–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Kd/10−8 M | ΔG°/kJ mol−1 |
|---|---|---|
| [Pd(phen)tu2]Cl2 | 6 | 40.8 |
| [Pd(phen)(nBut-tu)2]Cl2 | 9 | 39.8 |
| [Pt(phen)(nBut-tu)2]Cl2 | 20 | 37.8 |
| [Pt(phen)tu2]Cl2 | 40 | 36.1 |
| [Pt(bipy)(nBut-tu)2]Cl2 | 50 | 35.6 |
| [Pd(bipy)(nBut-tu)2]Cl2 | 100 | 33.9 |
| [Pd(bipy)tu2]Cl2 | 200 | 32.2 |
| [Pt(bipy)tu2]Cl2 | 280 | 31.3 |
| Drugs (5 µM) | 2008 Cells | C13* Cells | ||||||
|---|---|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | Apoptosis | G0/G1 | S | G2/M | Apoptosis | |
| Control | 66.9 ± 3 | 11.5 ± 1 | 14.6 ± 3.5 | 1.53 ± 1 | 62.7 ± 4 | 17.1 ± 2 | 13.6 ± 2 | 1.4 ± 0.2 |
| [Pd(bipy)(tu)2]Cl2 | 69.7 ± 3 | 12.1 ± 1 | 14.1 ± 0.2 | 1.24 ± 0.5 | 59.7 ± 4 | 19.8 ± 1 | 13.7 ± 1 | 2.1 ± 0.3 |
| [Pd(bipy)(Me-tu)2]Cl2 | 63.5 ± 5 | 20.5 ± 3 | 11.8 ± 1 | 1.44 ± 0.4 | 54.2 ± 2 | 22.7 ± 4 | 16.8 ± 2 | 3.4 ± 1.5 |
| [Pd(bipy)(nBu-tu)2]Cl2 | 73.5 ± 6 | 12.6 ± 3 | 12.8 ± 4 | 1.12 ± 0.2 | 67.8 ± 5 | 13.9 ± 2 | 15.3 ± 3 | 1.32 ± 0.4 |
| [Pd(bipy)(Et2-tu)2]Cl2 | 69.6 ± 5 | 12.7 ± 2 | 14.2 ± 3 | 1.30 ± 0.3 | 66.4 ± 7 | 14.9 ± 2 | 14.4 ± 3 | 1.37 ± 0.4 |
| [Pd(phen)tu2]Cl2 | 77.2 ± 2 | 3.2 ± 0.8 | 4.1 ± 0.2 | 9.8 ± 1.7 | 81.6 ± 2 | 2.3 ± 0.3 | 3.1 ± 0.3 | 10.6 ± 0.5 |
| [Pd(phen)(Me-tu)2]Cl2 | 71.8 ± 1 | 3.6 ± 1.4 | 4.6 ± 1.8 | 14.8 ± 0.7 | 81.1 ± 2 | 2.4 ± 0.5 | 3.4 ± 1.2 | 9.5 ± 2 |
| [Pd(phen)(nBu-tu)2]Cl2 | 64.7 ± 2 | 2.2 ± 0.1 | 7.2 ± 2.2 | 24.5 ± 1.1 | 71.1 ± 6 | 12.7 ± 2 | 5.1 ± 2 | 11.6 ± 2 |
| [Pd(phen)(Et2-tu)2]Cl2 | 73.3 ± 3 | 2.4 ± 0.9 | 5.6 ± 0.8 | 16.8 ± 0.6 | 78.6 ± 4 | 4.5 ± 0.6 | 3.6 ± 0.4 | 12.7 ± 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marverti, G.; Gozzi, G.; Lauriola, A.; Ponterini, G.; Belluti, S.; Imbriano, C.; Costi, M.P.; D’Arca, D. The 1,10-Phenanthroline Ligand Enhances the Antiproliferative Activity of DNA-Intercalating Thiourea-Pd(II) and -Pt(II) Complexes Against Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 6122. https://doi.org/10.3390/ijms20246122
Marverti G, Gozzi G, Lauriola A, Ponterini G, Belluti S, Imbriano C, Costi MP, D’Arca D. The 1,10-Phenanthroline Ligand Enhances the Antiproliferative Activity of DNA-Intercalating Thiourea-Pd(II) and -Pt(II) Complexes Against Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cell Lines. International Journal of Molecular Sciences. 2019; 20(24):6122. https://doi.org/10.3390/ijms20246122
Chicago/Turabian StyleMarverti, Gaetano, Gaia Gozzi, Angela Lauriola, Glauco Ponterini, Silvia Belluti, Carol Imbriano, Maria Paola Costi, and Domenico D’Arca. 2019. "The 1,10-Phenanthroline Ligand Enhances the Antiproliferative Activity of DNA-Intercalating Thiourea-Pd(II) and -Pt(II) Complexes Against Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cell Lines" International Journal of Molecular Sciences 20, no. 24: 6122. https://doi.org/10.3390/ijms20246122