Molecular Mechanisms That Define Redox Balance Function in Pathogen-Host Interactions—Is There a Role for Dietary Bioactive Polyphenols?

Abstract
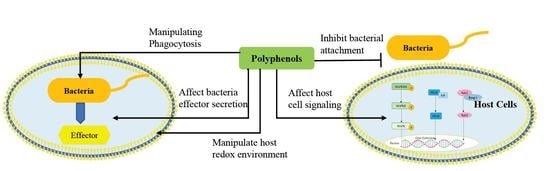
:
1. Introduction
2. Pathogen-Host Relationships
2.1. Bacterial Secretion Systems Facilitate Survival of Bacteria within the Host Cell
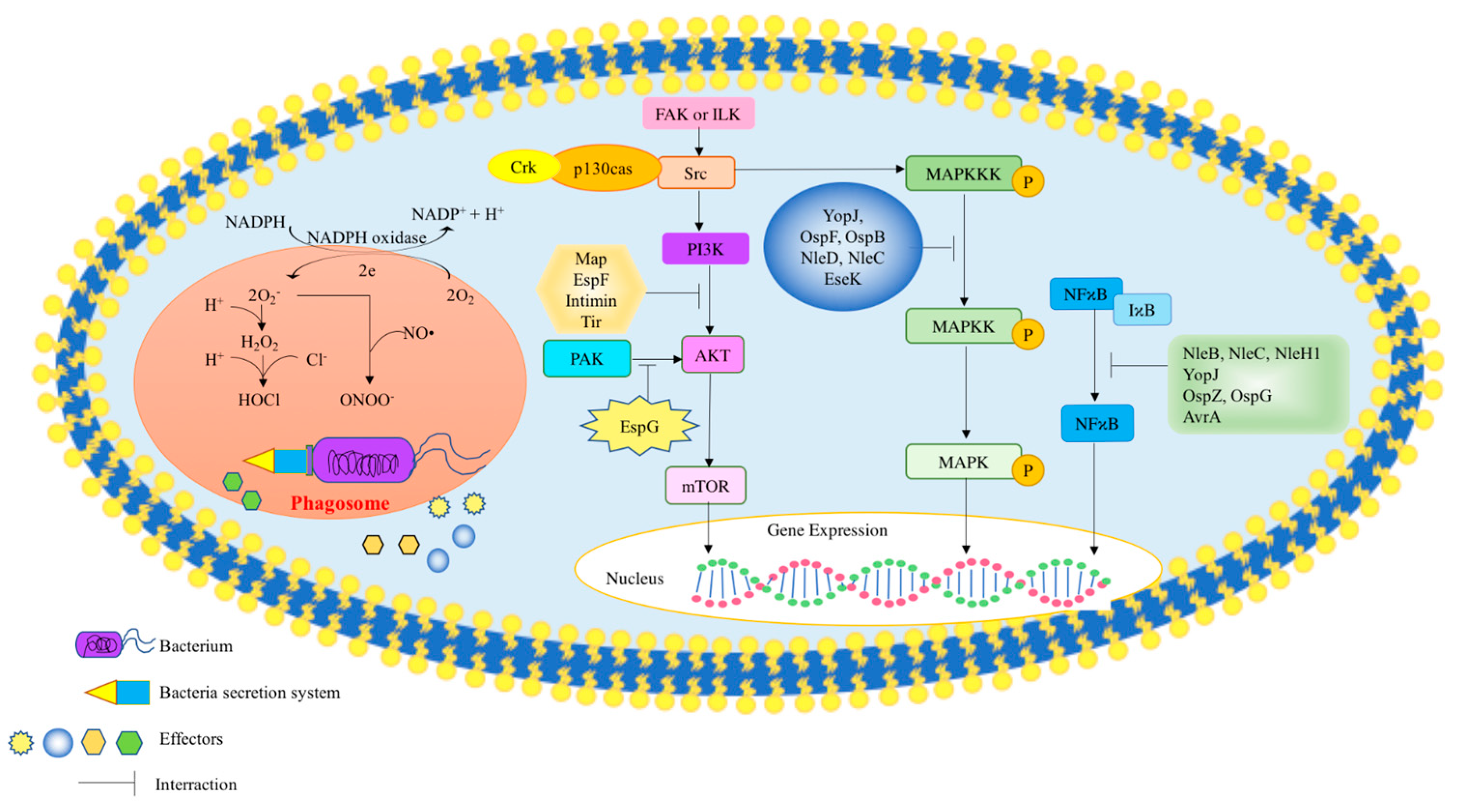
2.2. Mechanisms for Microbial Effectors to Influence Host Cell Signaling
2.2.1. MAPK (Mitogen-Activated Protein Kinases)
2.2.2. Phosphoinositide 3-Kinase (PI-3 Kinase)
2.2.3. Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κB)
2.2.4. p21 Activated Protein Kinase (PAK)
3. Disruption of Pathogen–Host Cell Interaction and Cell Signaling Pathways by Polyphenols
3.1. Effect of Polyphenol on Adhesion of Host Cells
3.2. Mechanisms by which Dietary Polyphenols Manipulate Host Redox Environment
3.3. Targeting Virulence of Pathogen by Manipulating Phagocytosis
3.4. Effect of Polyphenol on Bacterial Effector Secretion
3.5. Effect of Polyphenol on Host Cell Signaling
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABTS | 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid |
| ARE | AU-rich elements |
| BA | benzoic acid |
| CDC42 | cell division cycle 42 |
| cPLA2 | cytosolic phospholipase A2 |
| DNA | deoxyribonucleic acid |
| DNMT | DNA methyltransferases |
| DPPH | 1,1-diphenyl-2-picryhydrazyl |
| ECG | (-)-epicatechin-3-gallate |
| EHEC | Enterohemorrhagic Escherichia coli |
| ER | Estrogen receptor |
| ERK | Extracellular signal-regulated kinases |
| FAK | Focal adhesion kinase |
| GSH | Glutathione |
| Grx | Glutaredoxin |
| HLEC | Human lens epithelial cell |
| HP1& HP2 | Hydroperoxidase 1-Hydroperoxidase 2 |
| IL-8 | Interleukin 8 |
| ILK | Integrin-linked kinase |
| iNOS | nitrix oxide synthase |
| JNK | c-jun N-terminal kinases |
| LEE | Locus of enterocyte effacement |
| MAPK | Mitogen-activated protein kinase |
| MnSOD | Mn superoxide dismutase |
| NADPH | nicotinaminde adenine dinucleotide phosphate |
| NF-κB | Nuclear factor κB |
| PAK | p21-activated kinase |
| PCA | p-coumaric acid |
| PI3K | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PMN | polymorphonuclear |
| RNA | ribonucleic acid |
| RNS | Reactive nitrogen species |
| RPS3 | Ribosomal protein S3 |
| ROS | reactive oxygen species |
| TACO | tryptophan aspartate containing coat protein |
| T3SS | Type III secretion systems |
| T4SS | Type IV secretion systems |
| T6SS | Type VI secretion systems |
| TKI | Tyrosine kinase inhibitor |
| TKIS | Tyrosine kinase inhibitor sensitive |
| TKIR | Tyrosine kinase inhibitor resistant |
| TMCA | 4-methoxy-cinnamic acid |
| TNF-α | Tumor necrosis factor α |
| TRAF | TNF receptor-associated factor 2 |
| TSG | TNF-stimulated gene |
References
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 215–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azenabor, A.A.; Muili, K.; Akoachere, J.-F.; Chaudhry, A. Macrophage antioxidant enzymes regulate Chlamydia pneumoniae chronicity: Evidence of the effect of redox balance on host-pathogen relationship. Immunobiology 2006, 211, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dai, C.; Brown, K.; Rajendiran, E.; Makarenko, S.; Baker, J.; Ma, C.; Halder, S.; Montero, M.; Ionescu, V.A.; et al. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G39–G49. [Google Scholar] [CrossRef] [PubMed]
- Storz, G.; Tartaglia, L.A.; Farr, S.B.; Ames, B.N. Bacterial Defenses against Oxidative Stress. Trends Genet. 1990, 6, 363–368. [Google Scholar] [CrossRef]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Woolery, A.R.; Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Biol. 2011, 195, 1083–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baureder, M.; Reimann, R.; Hederstedt, L. Contribution of catalase to hydrogen peroxide resistance in Enterococcus faecalis. FEMS Microbiol. Lett. 2012, 331, 160–164. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways (Reprinted from Thiol Metabolism and Redox Regulation of Cellular Functions). Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef]
- Scalbert, A.; Williamson, G. Dietary Intake and Bioavailability of Polyphenols. J. Nutr. 2000, 130, 2073S–2085S. [Google Scholar] [CrossRef]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P.E. Polyphenols and human health: Prevention of disease and mechanisms of action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [Green Version]
- Reniere, M.L. Reduce, Induce, Thrive: Bacterial Redox Sensing during Pathogenesis. J. Bacteriol. 2018, 200, e00128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, J.K. What really happens in the neutrophil phagosome? Free Radic. Biol. Med. 2012, 53, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alto, N.M.; Orth, K. Subversion of Cell Signaling by Pathogens. Cold Spring Harb. Perspect. Biol. 2012, 4, a006114. [Google Scholar] [CrossRef] [PubMed]
- Riboulet, E.; Verneuil, N.; La Carbona, S.; Sauvageot, N.; Auffray, Y.; Hartke, A.; Giard, J.-C. Relationships between oxidative stress response and virulence in Enterococcus faecalis. J. Mol. Microbiol. Biotechnol. 2007, 13, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Von Ossowski, I.; Mulvey, M.R.; Leco, P.A.; Borys, A.; Loewen, P.C. Nucleotide sequence of Escherichia coli katE, which encodes catalase HPII. J. Bacteriol. 1991, 173, 514–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, L.B. Bacterial defenses against oxidants: Mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch. Biochem. Biophys. 2005, 433, 240–254. [Google Scholar] [CrossRef]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Ann. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [Green Version]
- Wan, B.; Zhang, Q.; Ni, J.; Li, S.; Wen, D.; Li, J.; Xiao, H.; He, P.; Ou, H.-Y.; Tao, J.; et al. Type VI secretion system contributes to Enterohemorrhagic Escherichia coli virulence by secreting catalase against host reactive oxygen species (ROS). PLoS Pathog. 2017, 13, e1006246. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.L.; Jechorek, R.P.; Erlandsen, S.L. Evidence for the translocation of Enterococcus faecalis across the mouse intestinal tract. J. Infect. Dis. 1990, 162, 82–90. [Google Scholar] [CrossRef]
- Diaz-Ochoa, V.E.; Lam, D.; Lee, C.S.; Klaus, S.; Behnsen, J.; Liu, J.Z.; Chim, N.; Nuccio, S.-P.; Rathi, S.G.; Mastroianni, J.R.; et al. Salmonella Mitigates Oxidative Stress and Thrives in the Inflamed Gut by Evading Calprotectin-Mediated Manganese Sequestration. Cell Host Microbe 2016, 19, 814–825. [Google Scholar] [CrossRef]
- Ochsner, U.A.; Vasil, M.L.; Alsabbagh, E.; Parvatiyar, K.; Hassett, D.J. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J. Bacteriol. 2000, 182, 4533–4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamet, A.; Kiss, E.; Batut, J.; Puppo, A.; Hérouart, D. The katA catalase gene is regulated by OxyR in both free-living and symbiotic Sinorhizobium meliloti. J. Bacteriol. 2005, 187, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Carbona, S.; Sauvageot, N.; Giard, J.-C.; Benachour, A.; Posteraro, B.; Auffray, Y.; Sanguinetti, M.; Hartke, A. Comparative study of the physiological roles of three peroxidases (NADH peroxidase, Alkyl hydroperoxide reductase and Thiol peroxidase) in oxidative stress response, survival inside macrophages and virulence of Enterococcus faecalis. Mol. Microbiol. 2007, 66, 1148–1163. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.G.; Hassett, D.J.; Stewart, P.S.; Schweizer, H.P.; McDermott, T.R. Protective role of catalase in Pseudomonas aeruginosa biofilm resistance to hydrogen peroxide. Appl. Environ. Microbiol. 1999, 65, 4594–4600. [Google Scholar]
- Kovacic, P.; Pozos, R.S. Cell signaling (mechanism and reproductive toxicity): Redox chains, radicals, electrons, relays, conduit, electrochemistry, and other medical implications. Birth Defects Res. C Embryo Today 2006, 78, 333–344. [Google Scholar] [CrossRef]
- Rosenberger, C.M.; Finlay, B.B. Phagocyte sabotage: Disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 2003, 4, 385–396. [Google Scholar] [CrossRef]
- Wan, M.; Zhou, Y.; Zhu, Y. Subversion of Macrophage Functions by Bacterial Protein Toxins and Effectors. Curr. Issues Mol. Biol. 2018, 25, 61–80. [Google Scholar] [CrossRef]
- Zhuang, X.; Chen, Z.; He, C.; Wang, L.; Zhou, R.; Yan, D.; Ge, B. Modulation of host signaling in the inflammatory response by enteropathogenic Escherichia coli virulence proteins. Cell. Mol. Immunol. 2017, 14, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Mattock, E.; Blocker, A.J. How Do the Virulence Factors of Shigella Work Together to Cause Disease? Front. Cell. Infect. Microbiol. 2017, 7, 64. [Google Scholar] [CrossRef]
- Ashida, H.; Sasakawa, C. Shigella hacks host immune responses by reprogramming the host epigenome. EMBO J. 2014, 33, 2598–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, C.; Pompili, M.; Scribano, D.; Limongi, D.; Petrucca, A.; Cannavacciuolo, S.; Schippa, S.; Zagaglia, C.; Grossi, M.; Nicoletti, M. The Shigella flexneri OspB effector: An early immunomodulator. Int. J. Med. Microbiol. 2015, 305, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossenkämper, A.; Marchès, O.; Fairclough, P.D.; Warnes, G.; Stagg, A.J.; Lindsay, J.O.; Evans, P.C.; Luong, L.A.; Croft, N.M.; Naik, S.; et al. Inhibition of NF-κB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J. Immunol. 2010, 185, 4118–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Han, F.; Tan, J.; Hou, M.; Zhang, Y.; Yang, D.; Liu, Q. Edwardsiella piscicida Type III Secretion System Effector EseK Inhibits Mitogen-Activated Protein Kinase Phosphorylation and Promotes Bacterial Colonization in Zebrafish Larvae. Infect. Immun. 2018, 86, 1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penuel, E.; Martin, G.S. Transformation by v-Src: Ras-MAPK and PI3K-mTOR mediate parallel pathways. Mol. Biol. Cell 1999, 10, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Krasilnikov, M.A. Phosphatidylinositol-3 kinase dependent pathways: The role in control of cell growth, survival, and malignant transformation. Biochem. Mosc. 2000, 65, 59–67. [Google Scholar]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar]
- Quitard, S.; Dean, P.; Maresca, M.; Kenny, B. The enteropathogenic Escherichia coli EspF effector molecule inhibits PI-3 kinase-mediated uptake independently of mitochondrial targeting. Cell. Microbiol. 2006, 8, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Guan, K.L.; Dixon, J.E. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science 1990, 249, 553–556. [Google Scholar] [CrossRef]
- Bliska, J.B.; Guan, K.L.; Dixon, J.E.; Falkow, S. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc. Natl. Acad. Sci. USA 1991, 88, 1187–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.S.; Bliska, J.B. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J. 1997, 16, 2730–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele-Mortimer, O.; Knodler, L.A.; Marcus, S.L.; Scheid, M.P.; Goh, B.; Pfeifer, C.G.; Duronio, V.; Finlay, B.B. Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J. Biol. Chem. 2000, 275, 37718–37724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, K.G.; Winfree, S.; Malik-Kale, P.; Jolly, C.; Ireland, R.; Knodler, L.A.; Steele-Mortimer, O. Activation of Akt by the bacterial inositol phosphatase, SopB, is wortmannin insensitive. PLoS ONE 2011, 6, e22260. [Google Scholar] [CrossRef] [Green Version]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef]
- Siomek, A. NF-κB signaling pathway and free radical impact. Acta Biochim. Pol. 2012, 59, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Gao, X.; Singh, G.; Hardwidge, P.R. Escherichia coli virulence protein NleH1 interaction with the v-Crk sarcoma virus CT10 oncogene-like protein (CRKL) governs NleH1 inhibition of the ribosomal protein S3 (RPS3)/nuclear factor κB (NF-κB) pathway. J. Biol. Chem. 2013, 288, 34567–34574. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: A KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, A.; Chernoff, J. Group I p21-activated kinases: Emerging roles in immune function and viral pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 13–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Molli, P.R.; Pakala, S.B.; Bui Nguyen, T.M.; Rayala, S.K.; Kumar, R. PAK thread from amoeba to mammals. J. Cell. Biochem. 2009, 107, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglieri, D.M.; Ushio-Fukai, M.; Monasky, M.M. P21-activated kinase in inflammatory and cardiovascular disease. Cell. Signal. 2014, 26, 2060–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- John Von Freyend, S.; Kwok-Schuelein, T.; Netter, H.J.; Haqshenas, G.; Semblat, J.-P.; Doerig, C. Subverting Host Cell P21-Activated Kinase: A Case of Convergent Evolution across Pathogens. Pathogens 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germane, K.L.; Spiller, B.W. Structural and functional studies indicate that the EPEC effector, EspG, directly binds p21-activated kinase. Biochemistry 2011, 50, 917–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selyunin, A.S.; Sutton, S.E.; Weigele, B.A.; Reddick, L.E.; Orchard, R.C.; Bresson, S.M.; Tomchick, D.R.; Alto, N.M. The assembly of a GTPase-kinase signalling complex by a bacterial catalytic scaffold. Nature 2011, 469, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Kamanova, J.; Lara-Tejero, M.; Galán, J.E. Salmonella stimulates pro-inflammatory signalling through p21-activated kinases bypassing innate immune receptors. Nat. Microbiol. 2018, 3, 1122–1130. [Google Scholar] [CrossRef]
- Churin, Y.; Kardalinou, E.; Meyer, T.F.; Naumann, M. Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in Helicobacter pylori infection. Mol. Microbiol. 2001, 40, 815–823. [Google Scholar] [CrossRef]
- Krautkrämer, E.; Giese, S.I.; Gasteier, J.E.; Muranyi, W.; Fackler, O.T. Human immunodeficiency virus type 1 Nef activates p21-activated kinase via recruitment into lipid rafts. J. Virol. 2004, 78, 4085–4097. [Google Scholar] [CrossRef] [Green Version]
- Filloux, A. Protein Secretion Systems in Pseudomonas aeruginosa: An Essay on Diversity, Evolution, and Function. Front. Microbiol. 2011, 2, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, H.J.; Pearson, J.S.; Badea, L.; Kelly, M.; Lucas, M.; Holloway, G.; Wagstaff, K.M.; Dunstone, M.A.; Sloan, J.; Whisstock, J.C.; et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 2010, 6, e1000898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creuzburg, K.; Giogha, C.; Wong Fok Lung, T.; Scott, N.E.; Mühlen, S.; Hartland, E.L.; Pearson, J.S. The Type III Effector NleD from Enteropathogenic Escherichia coli Differentiates between Host Substrates p38 and JNK. Infect. Immun. 2017, 85, e1000743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, H.P.; Shames, S.R.; Croxen, M.A.; Ma, C.; Chan, J.M.; Khan, M.A.; Wickham, M.E.; Deng, W.; Finlay, B.B.; Vallance, B.A. Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-κB and p38 mitogen-activated protein kinase activation. Infect. Immun. 2011, 79, 3552–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, H.; Ooka, T.; Iguchi, A.; Hayashi, T.; Sugimoto, N.; Tobe, T. NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 2010, 6, e1001231. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Muto, T.; Kleppe, M.; Bolanos, L.C.; Hueneman, K.M.; Walker, C.S.; Sampson, L.; Wellendorf, A.M.; Chetal, K.; Choi, K.; et al. TRAF6 Mediates Basal Activation of NF-κB Necessary for Hematopoietic Stem Cell Homeostasis. Cell Rep. 2018, 22, 1250–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, X.; Wu, X.-L.; He, L.-S.; Zeng, X.-F.; Crammer, A.C.; Lipsky, P.E. Competition between TRAF2 and TRAF6 regulates NF-kappaB activation in human B lymphocytes. Chin. Med. Sci. J. 2010, 25, 1–12. [Google Scholar] [CrossRef]
- Royan, S.V.; Jones, R.M.; Koutsouris, A.; Roxas, J.L.; Falzari, K.; Weflen, A.W.; Kim, A.; Bellmeyer, A.; Turner, J.R.; Neish, A.S.; et al. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-κB activation. Mol. Microbiol. 2010, 78, 1232–1245. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Nisan, I.; Yona, C.; Hanski, E.; Rosenshine, I. EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol. Microbiol. 2003, 47, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Bellmeyer, A.; Cotton, C.; Kanteti, R.; Koutsouris, A.; Viswanathan, V.K.; Hecht, G. Enterohemorrhagic Escherichia coli suppresses inflammatory response to cytokines and its own toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G576–G581. [Google Scholar] [CrossRef] [Green Version]
- Pha, K.; Navarro, L. Yersinia type III effectors perturb host innate immune responses. World J. Biol. Chem. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Von Pawel-Rammingen, U.; Telepnev, M.V.; Schmidt, G.; Aktories, K.; Wolf-Watz, H.; Rosqvist, R. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: A mechanism for disruption of actin microfilament structure. Mol. Microbiol. 2000, 36, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, H.; Zhou, Y.; Zhang, J.; Long, C.; Li, S.; Chen, S.; Zhou, J.-M.; Shao, F. The phosphothreonine lyase activity of a bacterial type III effector family. Science 2007, 315, 1000–1003. [Google Scholar] [CrossRef]
- Grishin, A.M.; Beyrakhova, K.A.; Cygler, M. Structural insight into effector proteins of Gram-negative bacterial pathogens that modulate the phosphoproteome of their host. Protein Sci. 2015, 24, 604–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, S. Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol. Chem. 2011, 392, 837–847. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Bolwell, P.G.; Bramley, P.M.; Pridham, J.B. The relative antioxidant activities of plant-derived polyphenolic flavonoids. Free Radic. Res. 1995, 22, 375–383. [Google Scholar] [CrossRef]
- Liang, N.; Kitts, D.D. Amelioration of Oxidative Stress in Caco-2 Cells Treated with Pro-inflammatory Proteins by Chlorogenic Acid Isomers via Activation of the Nrf2-Keap1-ARE-Signaling Pathway. J. Agric. Food Chem. 2018, 66, 11008–11017. [Google Scholar] [CrossRef]
- Liang, N.; Dupuis, J.H.; Yada, R.Y.; Kitts, D.D. Chlorogenic acid isomers directly interact with Keap 1-Nrf2 signaling in Caco-2 cells. Mol. Cell. Biochem. 2019, 457, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Boudet, A.-M. Evolution and current status of research in phenolic compounds. Phytochemistry 2007, 68, 2722–2735. [Google Scholar] [CrossRef]
- Cozens, D.; Read, R.C. Anti-adhesion methods as novel therapeutics for bacterial infections. Expert Rev. Anti Infect. 2012, 10, 1457–1468. [Google Scholar] [CrossRef]
- Asadi, A.; Razavi, S.; Talebi, M.; Gholami, M. A review on anti-adhesion therapies of bacterial diseases. Infection 2019, 47, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Orth, K. Targeting the bacteria-host interface: Strategies in anti-adhesion therapy. Virulence 2013, 4, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.M.; Kitts, D.D. Flavonoid composition of orange peel extract ameliorates alcohol induced tight junction dysfunction in Caco-2 monolayer. Food Chem. Toxicol. 2017, 105, 398–406. [Google Scholar] [CrossRef]
- Yamanaka, A.; Kimizuka, R.; Kato, T.; Okuda, K. Inhibitory effects of cranberry juice on attachment of oral streptococci and biofilm formation. Oral Microbiol. Immunol. 2004, 19, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pinzón-Arango, P.A.; Gallardo-Moreno, A.M.; Camesano, T.A. Direct adhesion force measurements between E. coli and human uroepithelial cells in cranberry juice cocktail. Mol. Nutr. Food Res. 2010, 54, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Toivanen, M.; Huttunen, S.; Lapinjoki, S.; Tikkanen-Kaukanen, C. Inhibition of adhesion of Neisseria meningitidis to human epithelial cells by berry juice polyphenolic fractions. Phytother. Res. 2011, 25, 828–832. [Google Scholar] [CrossRef] [Green Version]
- Furiga, A.; Lonvaud-Funel, A.; Dorignac, G.; Badet, C. In vitro anti-bacterial and anti-adherence effects of natural polyphenolic compounds on oral bacteria. J. Appl. Microbiol. 2008, 105, 1470–1476. [Google Scholar] [CrossRef]
- Esteban-Fernández, A.; Zorraquín-Peña, I.; Ferrer, M.D.; Mira, A.; Bartolomé, B.; González de Llano, D.; Moreno-Arribas, M.V. Inhibition of Oral Pathogens Adhesion to Human Gingival Fibroblasts by Wine Polyphenols Alone and in Combination with an Oral Probiotic. J. Agric. Food Chem. 2018, 66, 2071–2082. [Google Scholar] [CrossRef]
- Burger, O.; Weiss, E.; Sharon, N.; Tabak, M.; Neeman, I.; Ofek, I. Inhibition of Helicobacter pylori adhesion to human gastric mucus by a high-molecular-weight constituent of cranberry juice. Crit. Rev. Food Sci. Nutr. 2002, 42, 279–284. [Google Scholar] [CrossRef]
- Weiss, E.L.; Lev-Dor, R.; Sharon, N.; Ofek, I. Inhibitory effect of a high-molecular-weight constituent of cranberry on adhesion of oral bacteria. Crit. Rev. Food Sci. Nutr. 2002, 42, 285–292. [Google Scholar] [CrossRef]
- Parkar, S.G.; Stevenson, D.E.; Skinner, M.A. The potential influence of fruit polyphenols on colonic microflora and human gut health. Int. J. Food Microbiol. 2008, 124, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Hanasaki, Y.; Ogawa, S.; Fukui, S. The correlation between active oxygens scavenging and antioxidative effects of flavonoids. Free Radic. Biol. Med. 1994, 16, 845–850. [Google Scholar] [CrossRef]
- Hatano, T.; Yasuhara, T.; Yoshihara, R.; Agata, I.; Noro, T.; Okuda, T. Effects of interaction of tannins with co-existing substances. VII. Inhibitory effects of tannins and related polyphenols on xanthine oxidase. Chem. Pharm. Bull. 1990, 38, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Wachi, M.; Woo, J.-T.; Kato, M.; Kasai, S.; Takahashi, F.; Lee, I.-S.; Nagai, K. Fenton reaction is primarily involved in a mechanism of (-)-epigallocatechin-3-gallate to induce osteoclastic cell death. Biochem. Biophys. Res. Commun. 2002, 292, 94–101. [Google Scholar] [CrossRef]
- Nanjo, F.; Goto, K.; Seto, R.; Suzuki, M.; Sakai, M.; Hara, Y. Scavenging effects of tea catechins and their derivatives on 1,1-diphenyl-2-picrylhydrazyl radical. Free Radic. Biol. Med. 1996, 21, 895–902. [Google Scholar] [CrossRef]
- Afanas’ev, I.B.; Dorozhko, A.I.; Brodskii, A.V.; Kostyuk, V.A.; Potapovitch, A.I. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem. Pharmacol. 1989, 38, 1763–1769. [Google Scholar] [CrossRef]
- Miller, N.J.; Castelluccio, C.; Tijburg, L.; Rice-Evans, C. The antioxidant properties of theaflavins and their gallate esters—Radical scavengers or metal chelators? FEBS Lett. 1996, 392, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.E.; Khodr, H.; Hider, R.C.; Rice-Evans, C.A. Structural dependence of flavonoid interactions with Cu2+ ions: Implications for their antioxidant properties. Biochem. J. 1998, 33 Pt 3, 1173–1178. [Google Scholar] [CrossRef]
- Moran, J.F.; Klucas, R.V.; Grayer, R.J.; Abian, J.; Becana, M. Complexes of iron with phenolic compounds from soybean nodules and other legume tissues: Prooxidant and antioxidant properties. Free Radic. Biol. Med. 1997, 22, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Mira, L.; Fernandez, M.T.; Santos, M.; Rocha, R.; Florêncio, M.H.; Jennings, K.R. Interactions of flavonoids with iron and copper ions: A mechanism for their antioxidant activity. Free Radic. Res. 2002, 36, 1199–1208. [Google Scholar] [CrossRef]
- Lopes, G.K.; Schulman, H.M.; Hermes-Lima, M. Polyphenol tannic acid inhibits hydroxyl radical formation from Fenton reaction by complexing ferrous ions. Biochim. Biophys. Acta 1999, 1472, 142–152. [Google Scholar] [CrossRef]
- Akagawa, M.; Shigemitsu, T.; Suyama, K. Production of hydrogen peroxide by polyphenols and polyphenol-rich beverages under quasi-physiological conditions. Biosci. Biotechnol. Biochem. 2003, 67, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Abraham, T.E.; Zakaria, Z.A. Reactivity of phenolic compounds towards free radicals under in vitro conditions. J. Food Sci. Technol. 2015, 52, 5790–5798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozturk Sarikaya, S.B. Acethylcholinesterase inhibitory potential and antioxidant properties of pyrogallol. J. Enzym. Inhib. Med. Chem. 2015, 30, 761–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gülçin, I. Antioxidant activity of caffeic acid (3,4-dihydroxycinnamic acid). Toxicology 2006, 217, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, C.; Mo, H.; Ma, H.; Yuan, E.; Li, Q. Characterization and DPPH Radical Scavenging Activity of Gallic Acid-Lecithin Complex. Trop. J. Pharm. Res. 2014, 13, 1333–1338. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Francisco-Márquez, M.; Alvarez-Idaboy, J.R. Mechanism and kinetics studies on the antioxidant activity of sinapinic acid. Phys. Chem. Chem. Phys. 2011, 13, 11199–11205. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Kitts, D.D. Role of Chlorogenic Acids in Controlling Oxidative and Inflammatory Stress Conditions. Nutrients 2015, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Kono, Y.; Kobayashi, K.; Tagawa, S.; Adachi, K.; Ueda, A.; Sawa, Y.; Shibata, H. Antioxidant activity of polyphenolics in diets—Rate constants of reactions of chlorogenic acid and caffeic acid with reactive species of oxygen and nitrogen. Biochim. Biophys. Acta Gen. Subj. 1997, 1335, 335–342. [Google Scholar] [CrossRef]
- Nanjo, F.; Mori, M.; Goto, K.; Hara, Y. Radical scavenging activity of tea catechins and their related compounds. Biosci. Biotechnol. Biochem. 1999, 63, 1621–1623. [Google Scholar] [CrossRef] [Green Version]
- Cavia-Saiz, M.; Busto, M.D.; Pilar-Izquierdo, M.C.; Ortega, N.; Perez-Mateos, M.; Muñiz, P. Antioxidant properties, radical scavenging activity and biomolecule protection capacity of flavonoid naringenin and its glycoside naringin: A comparative study. J. Sci. Food Agric. 2010, 90, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Horváthová, K.; Novotný, L.; Tóthová, D.; Vachálková, A. Determination of free radical scavenging activity of quercetin, rutin, luteolin and apigenin in H2O2-treated human ML cells K562. Neoplasma 2004, 51, 395–399. [Google Scholar]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and prooxidant behavior of flavonoids: Structure-activity relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Hayakawa, F.; Kimura, T.; Maeda, T.; Fujita, M.; Sohmiya, H.; Fujii, M.; Ando, T. DNA cleavage reaction and linoleic acid peroxidation induced by tea catechins in the presence of cupric ion. Biochim. Biophys. Acta 1997, 1336, 123–131. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yamazaki, S.-I.; Kano, K.; Ikeda, T. Kinetic analysis and mechanistic aspects of autoxidation of catechins. Biochim. Biophys. Acta 2002, 1569, 35–44. [Google Scholar] [CrossRef]
- Miura, Y.H.; Tomita, I.; Watanabe, T.; Hirayama, T.; Fukui, S. Active oxygens generation by flavonoids. Biol. Pharm. Bull. 1998, 21, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Dashwood, W.-M.; Orner, G.A.; Dashwood, R.H. Inhibition of beta-catenin/Tcf activity by white tea, green tea, and epigallocatechin-3-gallate (EGCG): Minor contribution of H2O2 at physiologically relevant EGCG concentrations. Biochem. Biophys. Res. Commun. 2002, 296, 584–588. [Google Scholar] [CrossRef]
- Roques, S.C.; Landrault, N.; Teissèdre, P.-L.; Laurent, C.; Besançon, P.; Rouane, J.-M.; Caporiccio, B. Hydrogen peroxide generation in caco-2 cell culture medium by addition of phenolic compounds: Effect of ascorbic acid. Free Radic. Res. 2002, 36, 593–599. [Google Scholar] [CrossRef]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in cell culture: Rapid generation of hydrogen peroxide on addition of (-)-epigallocatechin, (-)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun. 2000, 273, 50–53. [Google Scholar] [CrossRef]
- Fujita, Y.; Wakabayashi, K.; Nagao, M.; Sugimura, T. Implication of hydrogen peroxide in the mutagenicity of coffee. Mutat. Res. 1985, 144, 227–230. [Google Scholar] [CrossRef]
- Alejandre-Durán, E.; Alonso-Moraga, A.; Pueyo, C. Implication of active oxygen species in the direct-acting mutagenicity of tea. Mutat. Res. 1987, 188, 251–257. [Google Scholar] [CrossRef]
- Gresham, H.D.; Lowrance, J.H.; Caver, T.E.; Wilson, B.S.; Cheung, A.L.; Lindberg, F.P. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 2000, 164, 3713–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaman, L.; Beaman, B.L. The Role of Oxygen and Its Derivatives in Microbial Pathogenesis and Host Defense. Ann. Rev. Microbiol. 1984, 38, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 2012, 5, pe10. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D. Cysteine 38 holds the key to NF-κB activation. Mol. Cell 2012, 45, 1–3. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, D.H.; Kim, D.G.; Lee, H.J.; Min, W.; Rhee, M.H.; Yun, B.S.; Kim, S. Phellinus baumii extract influences pathogenesis of Brucella abortus in phagocyte by disrupting the phagocytic and intracellular trafficking pathway. J. Appl. Microbiol. 2013, 114, 329–338. [Google Scholar] [CrossRef]
- Virgili, F.; Kobuchi, H.; Packer, L. Procyanidins extracted from Pinus maritima (Pycnogenol): Scavengers of free radical species and modulators of nitrogen monoxide metabolism in activated murine RAW 264.7 macrophages. Free Radic. Biol. Med. 1998, 24, 1120–1129. [Google Scholar] [CrossRef]
- Packer, L.; Rimbach, G.; Virgili, F. Antioxidant activity and biologic properties of a procyanidin-rich extract from pine (Pinus maritima) bark, pycnogenol. Free Radic. Biol. Med. 1999, 27, 704–724. [Google Scholar] [CrossRef]
- Anand, P.K.; Kaul, D.; Sharma, M. Green tea polyphenol inhibits Mycobacterium tuberculosis survival within human macrophages. Int. J. Biochem. Cell Biol. 2006, 38, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, A.; Li, J.; Zeng, Q.; Khokhani, D.; Hutchins, W.C.; Yost, A.C.; Biddle, E.; Toone, E.J.; Chen, X.; Yang, C.-H. Derivatives of plant phenolic compound affect the type III secretion system of Pseudomonas aeruginosa via a GacS-GacA two-component signal transduction system. Antimicrob. Agents Chemother. 2012, 56, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Peng, Q.; Selimi, D.; Wang, Q.; Charkowski, A.O.; Chen, X.; Yang, C.-H. The plant phenolic compound p-coumaric acid represses gene expression in the Dickeya dadantii type III secretion system. Appl. Environ. Microbiol. 2009, 75, 1223–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khokhani, D.; Zhang, C.; Li, Y.; Wang, Q.; Zeng, Q.; Yamazaki, A.; Hutchins, W.; Zhou, S.-S.; Chen, X.; Yang, C.-H. Discovery of plant phenolic compounds that act as type III secretion system inhibitors or inducers of the fire blight pathogen, Erwinia amylovora. Appl. Environ. Microbiol. 2013, 79, 5424–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Tian, F.; Li, J.; Hutchins, W.; Chen, H.; Yang, F.; Yuan, X.; Cui, Z.; Yang, C.-H.; He, C. Identification of phenolic compounds that suppress the virulence of Xanthomonas oryzae on rice via the type III secretion system. Mol. Plant Pathol. 2017, 18, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ding, W.; Zhang, Y.; Liu, X.; Yang, L. Oleanolic Acid Induces the Type III Secretion System of Ralstonia solanacearum. Front. Microbiol. 2015, 6, 1466. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M. Cellular targets for the beneficial actions of tea polyphenols. Am. J. Clin. Nutr. 2013, 98, 1642S–1650S. [Google Scholar] [CrossRef] [Green Version]
- Das, J.; Ramani, R.; Suraju, M.O. Polyphenol compounds and PKC signaling. Biochim. Biophys. Acta 2016, 1860, 2107–2121. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Phan, A.N.H.; Choi, J.-W. Anti-cancer Effects of Polyphenolic Compounds in Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-resistant Non-small Cell Lung Cancer. Pharm. Mag. 2017, 13, 595–599. [Google Scholar]
- Xie, Q.; Bai, Q.; Zou, L.-Y.; Zhang, Q.-Y.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.-D.; Mi, M.-T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014, 53, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yu, R.; Owuor, E.D.; Kong, A.N. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch. Pharm. Res. 2000, 23, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Torre, E. Molecular signaling mechanisms behind polyphenol-induced bone anabolism. Phytochem. Rev. 2017, 16, 1183–1226. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Bacteria | Effector | Reported Action | Reference |
|---|---|---|---|
| EHEC | Catalase KatN | Hydrolyze ROS | [18] |
| Enterococcus faecalis | KatA | Survive oxidative burst | [19] |
| Salmonella spp. | SodA and KatN | Detoxify ROS | [20] |
| Pseudomonas aeruginosa | Catalase KatB-AnkB, AhpB, and AhpC-AhpF | OxyR-dependent regulation against oxidative stress | [21] |
| Sinorhizobium meliloti | Catalase HPII, KatA and KatC | Decrease ROS concentration | [22] |
| Bacteria | Effectors | Cell Signaling Mechanisms | References |
|---|---|---|---|
| E. coli | Type III effectors NleE and NleB protein | Down-regulate NF-κB by decreasing IKK phosphorylation | [34,62] |
| NleD (metalloprotease) | Inactivate JNK and p38. | [63] | |
| NleC (zinc protease) | Suppress inflammatory response by inactivating NF-κB and p38 | [64,65] | |
| Tir | Suppress both TRAF2 and TRAF6-induced NF-κB activation | [66,67] | |
| NleH1 | Suppress RPS3-induced NF-κB activation | [49,68] | |
| NleH2 | Suppress NleH1 | [68] | |
| EspH | Modulator of host cell actin cytoskeleton | [69] | |
| Locus of enterocyte effacement (LEE) encoded Map, EspF, Tir and Intimin proteins | Inhibit phosphorylation of Pl-3 kinase substrate | [39] | |
| EHEC | Stx2 toxin | Pro-inflammatory protein, promote IL-8 production. | [70] |
| EspG | Inhibit PAK signaling | [56,57] | |
| Yersinia | YopJ | Anti-inflammatory activity that inhibit both MAPK and NF-κB pathways | [33] |
| YopH | Inhibit Ca signaling and ROS production | [71] | |
| YopE (GTPase-activating protein) | Downregulate Rho, Rac and Cdc42 activity | [72] | |
| Shigella spp. | OspF (Phosphothreonine lyase) | Inactivate MAPK | [73] |
| OspB (Induces phosphorylation) | Activate MAPK | [30] | |
| OspZ | Inhibit NF-κB | [62] | |
| OspG | Stabilize IkB | [74] | |
| Salmonella spp. | AvrA (deubiquitinase) | Remove ubiquitin from IκB-alpha and beta-catenin | [48] |
| E. piscicida | EseK | Inhibit MAPK | [35] |
| Phenolic Compounds | Free Radical Scavenging Activity | Reference |
|---|---|---|
| Protocatechuic acid | Best against DPPH• and O2•− | [103] |
| Pyrogallol | Best against DPPH• and O2•−, effective against ABTS, DMPD, H2O2 | [104] |
| Caffeic acid | Best against DPPH• and O2•− | [105] |
| Gallic acid | Best against DPPH• and O2•− | [106] |
| Sinapinic acid | Hydroxyl radical scavenging | [107] |
| Chlorogenic acid | Hydroxyl radical and O2•− | [108,109] |
| Epicatechin | DPPH• scavenging, hydroxyl radical, and superoxide anion radical-scavenging activities | [110] |
| Naringenin | Hydroxyl and superoxide radical scavenger | [111] |
| Luteoloside | Against H2O2 radicals | [112] |
| Apigenin | Against H2O2 radicals and DPPH• scavenging | [112] |
| Polyphenols | Food Sources | Signaling Pathway | Mechanism | References |
|---|---|---|---|---|
| Catechin, theaflavin, and thearubigin | Tea products | Membrane intracellular receptors | Activation of cellular receptors which modifies intracellular signaling. Directly bind to ribonucleic acid (RNA) and deoxyribonucleic acid (DNA), allowing them to induce gene transcription. Assist in nuclear translocation of other transcription factors. Interact with transcription activator or repressor in the nucleus, altering gene expression. | [138] |
| Curcumin | Ginger | Interferes protein kinase C (PKC) signaling pathway, oncogenes and coding proteins | Regulates the transcription of the antioxidant enzyme genes through PKC signaling. Curcumin suppresses the activity of PKC which indirectly affecting MAPK and C-jun. | [139] |
| Equol, kaemferol, resveratrol, ellagic acid | Found in vegetables such as spinach, kale and endive. | Tyrosine kinase | Inhibits HCC827 panel, tyrosine kinase inhibitor (TKI)-sensitive (TKIS) and TKI-resistant clones. | [140] |
| Gallic acid, p-courmaric, heaperidin | Bark, wood, leaf, fruit, root and seed. Present in berries, plums, grapes, mango, tea, wine | Tyrosine kinase | Inhibit only tyrosine kinase inhibitor (TKI)-resistant TKIR cells H1993. | [140] |
| Genistein | Soy-based products such as chickpeas, tofu, soymilk, soy flour, soy protein, miso, tempeh | DNA methylation and histone modification. | Decreases DNA methylation of various tumor suppressor genes. Demethylate and reactivate TNF-stimulated gene (TSG), causing anticancer effect. DNA methylation is a critical part of transcriptional regulation that is catalyzed by specific DNA methyltransferases (DNMTs). Genistein alters histone to promote or prevent DNA replication. | [141] |
| Green Tea polyphenol (-)-epicatechin-3-gallate (ECG) | Green Tea | Activation of ERK and p38 | Induced AU-rich elements (ARE)-mediated gene expression to activate MAPK pathway, stimulate caspase-3 activity and induce apoptosis Anti-cancer and tumor property comes from their ability to suppress cellular growth and initiate apoptosis. | [142] |
| Phytoestrogens | Soy products and foods such as soybeans, tofu, miso, tempeh, vegetables, fruits, grains and legumes. | Target classical estrogen receptors (ER) pathway, TNF signaling pathway, and non-genomic signaling | Binds to ERα or ERβ, and induces estrogen receptor expression (ERE)-dependent transcription. Inhibiting tumor necrosis factor-α (TNF-α)-induced apoptosis. Involvement in pathways allow phenolic compounds to cross-talk to other transduction signals and a wide application potential. Activates estrogen pathway to regulate the expression gene responsible for the maintenance of bone mass. Thus phytoestrogens help balance bone resorption and bone formation. | [143] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, K.; Wang, D.; Kitts, D.D. Molecular Mechanisms That Define Redox Balance Function in Pathogen-Host Interactions—Is There a Role for Dietary Bioactive Polyphenols? Int. J. Mol. Sci. 2019, 20, 6222. https://doi.org/10.3390/ijms20246222
Mu K, Wang D, Kitts DD. Molecular Mechanisms That Define Redox Balance Function in Pathogen-Host Interactions—Is There a Role for Dietary Bioactive Polyphenols? International Journal of Molecular Sciences. 2019; 20(24):6222. https://doi.org/10.3390/ijms20246222
Chicago/Turabian StyleMu, Kaiwen, Danni Wang, and David D. Kitts. 2019. "Molecular Mechanisms That Define Redox Balance Function in Pathogen-Host Interactions—Is There a Role for Dietary Bioactive Polyphenols?" International Journal of Molecular Sciences 20, no. 24: 6222. https://doi.org/10.3390/ijms20246222